
| Spécialité | Pneumologie |
|---|
| CIM-10 | J84.1 |
|---|---|
| CIM-9 | 516.3 |
| OMIM | 178500 |
| DiseasesDB | 4815 |
| MedlinePlus | 000069 |
| eMedicine | 363273 |
| MeSH | D011658 |
| Patient UK | Idiopathic-pulmonary-fibrosis |
La fibrose pulmonaire idiopathique (FPI) est une maladie chronique, dévastatrice et mortelle, caractérisée par la dégradation progressive des fonctions pulmonaires. L'expression « fibrose pulmonaire » fait référence à une cicatrisation des tissus pulmonaires provoquant une dyspnée (difficulté à respirer) qui s’aggrave avec le temps. La fibrose est souvent associée à un mauvais pronostic. Le mot « idiopathique » signifie que la cause de cette fibrose est encore inconnue.
La fibrose pulmonaire idiopathique concerne principalement les adultes âgés de 50 à 70 ans mais elle peut aussi subvenir plus tôt, même chez les jeunes de moins de 20 ans et les jeunes enfants , plus particulièrement ceux ayant des antécédents de tabagisme. Les hommes sont plus touchés que les femmes. Près de 17 000 personnes en France et 3 millions de personnes dans le monde ont cette maladie.
La fibrose pulmonaire idiopathique appartient à la famille des pneumopathies interstitielles, composée de plus de 200 maladies touchant l’interstitium. Ce tissu, situé entre les alvéoles pulmonaires, est le principal site des lésions provoquant les pneumopathies interstitielles ; mais ces lésions touchent également souvent les espaces alvéolaires, les voies aériennes périphériques et les vaisseaux sanguins. Le tissu pulmonaire présente alors un schéma histopathologique caractéristique, de pneumopathie interstitielle usuelle. La pneumopathie interstitielle usuelle a donc par les mêmes manifestations pathologiques que la fibrose pulmonaire idiopathique.
En 2011, de nouvelles recommandations pour le diagnostic et la prise en charge de la fibrose pulmonaire idiopathique ont été publiées. Le diagnostic de la fibrose pulmonaire idiopathique ne peut être posé qu’après avoir écarté toutes les causes connues de pneumopathie interstitielle, et avoir vérifié la présence d’un modèle radiologique spécifique grâce à une tomodensitométrie haute résolution. Dans un contexte clinique adapté, une fibrose pulmonaire idiopathique peut être diagnostiquée par tomodensitométrie uniquement, sans avoir à recourir à une biopsie du poumon par voie chirurgicale.
Classification
La FPI (fibrose pulmonaire idiopathique) est l'une des 200 maladies pulmonaires de la famille des pneumopathies interstitielles (PI), qui touchent l'interstitium pulmonaire, le tissu situé entre les espaces alvéolaires des poumons. C'est une pneumopathie interstitielle idiopathique, qui est à son tour une pneumopathie interstitielle, aussi connue comme pneumopathie parenchymateuse diffuse.
La classification de 2002 des PPI établie par l'American Thoracic Society et l'European Respiratory Society (ATS/ERS) a été actualisée en 2013. Elle comporte maintenant trois grandes classes de PII :
- les PII majeures, qui regroupent les pneumopathies interstitielles chroniques fibrosantes (dont la FPI et la pneumonie interstitielle non spécifique [PINS]), les pneumopathies interstitielles dues au tabac (dont la pneumopathie interstitielle idiopathique - bronchiolite respiratoire [BR-PII], la pneumopathie interstitielle desquamante [DIP], les PI aiguë/subaiguë (dont la pneumonie organisée cryptogénique [POC] et la pneumopathie interstitielle aigüe [PIA].
- les PII rares
- les PII inclassables.
Le diagnostic de PII est posé après l'élimination de toutes les causes de pneumopathie interstitielle connues. Parmi les pneumopathies interstitielles dont les causes sont connues on peut citer la pneumopathie d'hypersensibilité, l’histiocytose langerhansienne, l’asbestose et la maladie du collagène vasculaire. Cependant souvent les lésions ne touchent pas seulement l’interstitium, mais aussi les voies supérieures, périphériques et les vaisseaux sanguins.
Le tableau suivant présente la nouvelle classification des PII.

Épidémiologie
Bien que rare, la fibrose pulmonaire idiopathique est la pneumopathie interstitielle idiopathique la plus courante.
Sa prévalence est estimée comprise entre 14,0 et 42,7 pour 100 000, selon une analyse des données du système de santé, avec des variations liées à la définition du cas utilisée dans l’analyse.
Elle est plus répandue chez les hommes que chez les femmes et généralement diagnostiquée chez les plus de 50 ans.
L’incidence de la fibrose pulmonaire idiopathique reste difficile à déterminer car tous les praticiens n’utilisent pas les mêmes critères de diagnostic. Dans les 28 pays de l’Union Européenne, une série de sources estime que l’incidence est comprise entre 4,6 et 7,4 pour 100 000, ce qui implique qu’entre 30 000 et 35,000 nouveaux cas environ sont diagnostiqués chaque année.
Une étude de cohorte observationnelle rétrospective monocentrique, menée au Danemark au Aarhus University Hospital entre 2003 et 2009, qui incluait des patients ayant été incidemment diagnostiqués atteints d'une PI indique que 4,1 pour 1,000 habitants développent chaque année une pneumopathie interstitielle. La fibrose pulmonaire idiopathique s'avére la pneumopathie la plus fréquente (28 %), suivie par la pneumopathie interstitielle liée à des troubles du tissu conjonctif (14 %), la pneumopathie d'hypersensibilité (7 %) et la pneumonie interstitielle non spécifique (PINS) (7 %). La fréquence des fibroses pulmonaires idiopathiques s'élève à 1,3 pour 1 000 habitants/année.
L'incidence semble être un peu plus faible en Asie ou en Amérique du Sud. Elle semble augmenter avec le temps.
Dans la mesure où, dans les pays européens, la distribution de la maladie est hétérogène il convient de revoir les données épidémiologiques dans le cadre d'un registre européen étendu répertoriant les PI et les FPI.
Causes/facteurs de risque de FPI
La FPI ou fibrose pulmonaire idiopathique est par définition une maladie idiopathique (c'est-à-dire dont on ignore la cause ou l'explication causale) ; des facteurs environnementaux et certaines expositions accroissent cependant le risque d'en développer.
Parmi les facteurs de risque environnementaux et/ou professionnels figurent :
- le tabagisme, qui à lui seul double le risque de développer une FPI ;
- l'exposition à certaines formes de pollution de l'air, particulaire notamment : exposition aux microparticules et poussières métalliques (qu'on trouve aussi dans les fumées de soudure), aux poussières de bois, de charbon, de pierre et d'autres matériaux riches en silice tels que bétons et granulats routier notamment (L'ANSES a conclu en que l'exposition par inhalation à des particules fines de silice est une source de risque pour cette maladie (et d'autres maladies autoimmunes, cancers, etc. ) ;
- les métiers des secteurs agricoles et d'élevage ;
- l'exposition à certains virus pourraient causer une fibrose pulmonaire idiopathique (et/ou d'autres maladies pulmonaires fibrosantes).
- de fait d'avoir développé une sarcoïdose (favorisée par certains métiers).
Cependant, certains patients semblent ne pas avoir été exposé à ces risques, qui donc ne suffisent pas à expliquer de façon exhaustive et satisfaisante la présence de la maladie.
Une prévalence accrue de Reflux gastro-œsophagien (RGO) est notée chez ces malades ; ce reflux pourrait éventuellement être un facteur étiologique de certaines FPI, ce qui reste à démontrer.
La cause génétique est rare ou absente, car un antécédent familial n'est trouvé que dans moins de 5% des cas de fibrose pulmonaire idiopathique ; et sur le plan clinique et histologique, il est impossible de déterminer si la maladie est liée aux antécédents ou non. Les associations génétiques incluent des mutations des protéines A1, A2, C (SFTPA1, SFTPA2B) du surfactant pulmonaire et des mucines (MUC5B). Un variant du gène MUC5B a été détecté chez environ 20 % des sujets d'origine européenne, (pays du nord et de l'ouest de l'Europe) et chez 19 % de la population de l'étude Framingham Heart Study sur les maladies cardiovasculaires. Cette mutation implique une surexpression de la mucine dans les bronches distales. Les mutations des gènes par les télomérases sont aussi associées aux antécédents de fibrose pulmonaire et à la fibrose pulmonaire idiopathique isolée chez certains patients (TERT, TERC). Une mutation liée au chromosome X du troisième composant de la télomérase, qui correspond à la dyskérine (DKC1) a été décrite chez une famille atteinte de FPI.
Physiopathologie
La fibrose pulmonaire idiopathique serait provoquée par un processus anormal de cicatrisation qui inclut ou provoque un dépôt excessif de collagène (fibrose) dans l’interstitium pulmonaire, avec une inflammation minime associée.
On suppose que les lésions initiales ou répétées touchent les cellules pulmonaires portant le nom de cellules épithéliales alvéolaires, qui couvrent la plus grande partie de la surface alvéolaire. Lorsque les cellules épithéliales alvéolaires de type I seraient endommagées ou détruites, les cellules épithéliales alvéolaires de type II prolifèreraient afin de couvrir la membrane basale exposée. Dans un cas normal, les cellules de type II hyperplasiques meurent et les cellules restantes se répandent et subissent un processus de différenciation afin de se transformer en cellules de type I. Dans un cas pathologique, et en présence de TGFβ (transforming growth factor beta), les fibroblastes s’accumulent là où les cellules ont été endommagées et se différencient en myofibroblastes qui sécrètent du collagène et d’autres protéines Autrefois, on pensait que l’inflammation était la cause de la cicatrisation des tissus. Cependant, selon les découvertes récentes, le développement des foyers fibroblastiques précède l’accumulation des cellules inflammatoires et le dépôt de collagène qui s’ensuit.
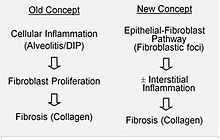
Ce modèle pathogénétique est indirectement soutenu par les caractéristiques cliniques de la fibrose pulmonaire idiopathique, notamment une déclaration insidieuse, une progression sur plusieurs années, une aggravation soudaine assez rare, et une absence de réponse à un traitement immunosuppresseur. Actuellement, plusieurs traitements ciblant l’activation du fibroblaste ou la synthèse de la matrice extracellulaire sont en cours d’essais ou de développement.
Le rôle des infections a également été suggéré sur plusieurs arguments : modification du microbiote pulmonaire, rôle d'une mutation du gène TOLLIP dont la protéine intervient dans la défense anti-microbienne.
Elle implique des protéases à sérine de la famille des Type II Transmembrane Serine Proteases (protéase transmembranaire à sérine 2) ; selon Awen Menou (2017) « l'activation de la cascade de la coagulation et des protéases à sérine, délétère dans la progression des maladies pulmonaires chroniques, est une caractéristique de la pathologie. » Récemment, un lien a été démontré entre Protease-Activated Receptor-2 (PAR-2), un récepteur cellulaire ubiquitaire, et la progression de la fibrose pulmonaire chez l'homme et la souris}. Or des facteurs de la coagulation peuvent activer le récepteur PAR-2, mais au moins deux protéines à sérine (protéases appartenant à une famille récemment identifiée : dite Type II Transmembrane Serine Proteases ou TTSPs) peuvent aussi le faire : ce sont la matriptase et la Human Airway Trypsin-like protease (HAT). xx
Awen Menou a récemment (2017) montré que dans les cas de FPI, l'expression et de l'activité de ces protéases de la famille des TTSPs était dérégulée et qu'in vitro, la matriptase provoque des réponses « pro-fibrosantes dans les fibroblastes pulmonaires primaires humains via l'activation de PAR-2, tandis que la HAT induit des réponses anti-fibrosantes dans ces cellules et une activation de la voie de la prostaglandine E2 ». De plus, in vivo, « l'inhibition génétique et pharmacologique de la matriptase atténue la fibrose dans le modèle murin de fibrose pulmonaire induite par la bléomycine, et des résultats similaires sont observés à la suite de la surexpression de la HAT médiée par adénovirus dans ce modèle animal ».
La découverte de cette implication de la matriptase et de la HAT, dans la pathogénèse de la FPI ouvre de nouvelles portes pour la recherche de médicaments.
Diagnostic
Un diagnostic précoce de la fibrose pulmonaire idiopathique est indispensable pour envisager le traitement et éventuellement l’amélioration clinique à long terme de cette maladie progressive et mortelle. En cas de suspicion de fibrose pulmonaire idiopathique, le diagnostic peut être délicat mais grâce à une approche multidisciplinaire, impliquant pneumologue, radiologue et spécialiste des pneumopathies interstitielles, la fibrose pulmonaire idiopathique peut être bien diagnostiquée.
En l'an 2000, une classification de consensus international multidisciplinaire de l’American Thoracic Society (ATS) et de l’European Respiratory Society (ERS) a proposé des critères, classés en principaux et secondaires, afin de diagnostiquer la fibrose pulmonaire idiopathique. Cependant, en 2011, de nouveaux critères pour le diagnostic et la prise en charge de la fibrose pulmonaire idiopathique, simplifiés et mis à jour, ont été publiés par l’ATS et l’ERS, avec l’aide de la Japanese Respiratory Society (JRS) et de la Latin American Thoracic Association (ALAT). Actuellement, pour diagnostiquer une fibrose pulmonaire idiopathique, il faut :
- exclure toute autre cause connue de pneumopathie idiopathique, comme une exposition à des facteurs environnementaux lors d’activités professionnelles ou privées, des troubles des tissus conjonctifs, ou une exposition/toxicité médicamenteuse ;
- avoir un aspect de pneumopathie interstitielle usuelle à la tomodensitométrie (ou scanner).
Dans un contexte clinique adapté, une fibrose pulmonaire idiopathique peut être diagnostiquée par tomodensitométrie seule, sans avoir à recourir à une biopsie du poumon.
Il peut être difficile de reconnaître une fibrose pulmonaire idiopathique dans la pratique clinique car les symptômes ressemblent souvent à ceux de pathologies plus courantes comme l’asthme, la broncho-pneumopathie chronique obstructive ou l’insuffisance cardiaque congestive. Il est capital que les cliniciens se demandent si les antécédents, symptômes (ou signes cliniques), examens radiologiques et explorations fonctionnelles respiratoires convergent tous vers un diagnostic de fibrose pulmonaire idiopathique ou si les résultats doivent conduire à un autre diagnostic. On a longtemps estimé qu’il était difficile de distinguer les patients atteints de pneumopathie interstitielle liée à une exposition à l’amiante ou à des médicaments (agents chimiothérapeutiques ou nitrofurantoïne), atteints de polyarthrite rhumatoïde et atteints de sclérodermie/sclérose systémique, de ceux atteints de fibrose pulmonaire idiopathique. La fibrose pulmonaire idiopathique peut aussi être confondue avec les pneumopathies interstitielles liées à des troubles des tissus conjonctifs, une sarcoïdose avancée, une pneumopathie d’hypersensibilité, une histiocytose Langerhansienne, ou une fibrose radio-induite.
Caractéristiques cliniques

| Fichier audio | |
| Râles crépitants | |
|
Râles crépitants de type Velcro à l’auscultation d’un patient atteint de fibrose pulmonaire idiopathique
|
|
|
| |
|---|---|
|
modifier |
Chez de nombreux patients, les symptômes se manifestent bien avant le diagnostic. Les signes cliniques les plus courants de la fibrose pulmonaire idiopathique incluent :
- âge (plus de 50 ans) ;
- toux sèche, non productive à l’effort ;
- aggravation de la dyspnée à l’effort (difficulté à respirer lors d’un exercice physique) ;
- à l’auscultation, râles crépitants inspiratoires secs bilatéraux des bases, de type « Velcro » (râles crépitants dans les poumons à l’inhalation, qui s’entendent au stéthoscope, comme si on arrachait lentement un Velcro) ;
- déformation des doigts, ou des orteils (hippocratisme digital);
- résultat des explorations fonctionnelles respiratoires anormaux, avec des échanges gazeux limités et de mauvaise qualité.
Ces caractéristiques sont dues à un déficit en oxygène dans le sang (hypoxémie) ; elles peuvent être associées à différents troubles pulmonaires et ne sont pas spécifiques à la fibrose pulmonaire idiopathique. Cependant la fibrose pulmonaire idiopathique doit être envisagée chez tous les patients ayant une dyspnée chronique inexpliquée à l’effort, ceux présentant une toux avec râles crépitants bilatéraux des bases à l’inspiration ou des doigts en forme de « baguettes de tambour ».
L’évaluation du râle crépitant de type « velcro » à l’auscultation permet facilement d’améliorer le diagnostic précoce de la fibrose pulmonaire idiopathique : un râle crépitant léger est facilement reconnaissable et constitue un signe clinique de la fibrose pulmonaire idiopathique.
Si de légers râles crépitants bilatéraux se font entendre tout le long de l’inspiration et persistent après plusieurs inspirations profondes, et s’ils persistent plusieurs semaines chez un sujet ≥ 60 ans, il y a suspicion de fibrose pulmonaire idiopathique et il faut envisager une tomodensitométrie haute résolution, plus précise qu’une simple radiographie du thorax. Dans la mesure où le râle crépitant n’est pas spécifique à la fibrose pulmonaire idiopathique, il doit déclencher un processus de recherche de diagnostic.
Si ces symptômes sont identifiés chez un patient, les recherches doivent être approfondies avant d’établir avec certitude le diagnostic de fibrose pulmonaire idiopathique.
Radiologie

La radiographie du thorax est utile pour le suivi courant des patients atteints de fibrose pulmonaire idiopathique. Une radiographie classique ne permet malheureusement pas d’établir le diagnostic mais peut faire état d’un volume pulmonaire diminué, avec des atteintes interstitielles réticulaires typiques importantes près de la base des poumons.
L’évaluation radiologique par tomodensitométrie haute résolution est capitale dans le diagnostic de la fibrose pulmonaire idiopathique. La tomographie axiale calculée par ordinateur est réalisée à l’aide d’un scanner, sans injection d’agents de contraste. Les coupes sont très fines (1–2 mm).
La tomodensitométrie haute résolution typique en cas de fibrose pulmonaire idiopathique fait apparaître des altérations fibrotiques des deux poumons, en particulier à la base et en périphérie. Selon les recommandations conjointes de l’ATS, l’ERS, la JRS et l’ALAT de 2001, la tomodensitométrie haute résolution est une composante essentielle du diagnostic de fibrose pulmonaire idiopathique, qui permet d’identifier la pneumopathie interstitielle usuelle grâce à la présence de signes suivants à répartition typiquement basale et périphérique bien que souvent inégale:
- opacités réticulaires, souvent associées à une bronchiectasie par traction ;
- images en nids d’abeilles se manifestant sous forme de cavités kystiques, en général de diamètres comparables (3–10 mm) mais souvent assez grandes, sous-pleurales et caractérisées par des parois bien définies et disposées en au moins deux rangées (La présence d’une seule rangée de kystes ne suffit pas à évoquer des nids d’abeille) ;
- opacités en verre dépoli, courantes mais moins répandues que la réticulation ;
- la répartition est typiquement basale et périphérique bien que souvent inégale.

Histologie
Selon les recommandations de 2011, en l’absence d’un schéma de pneumopathie interstitielle usuelle à la tomodensitométrie haute résolution, il faut réaliser des biopsies du poumon par voie chirurgicale pour confirmer le diagnostic.
Les spécimens histologiques pour le diagnostic de la fibrose pulmonaire idiopathique doivent être prélevés à au moins trois endroits différents et être assez importants pour que le pathologiste puisse observer la structure sous-jacente du poumon. Elles doivent éviter les zones les plus atteintes (radiologiquement) car on peut se retrouve alors avec une fibrose non spécifique. Les petites biopsies, comme celles obtenues par biopsie pulmonaire par voie transbronchique (réalisée durant une bronchoscopie) ne sont généralement pas suffisantes. Par conséquent, il faut généralement des biopsies plus importantes, réalisées par thoracotomie ou thoracoscopie.
Le tissu pulmonaire chez les patients atteints de fibrose pulmonaire idiopathique se caractérise généralement par un modèle histopathologique de pneumopathie interstitielle usuelle et se caractérise donc par les mêmes manifestations pathologiques que la fibrose pulmonaire idiopathique.
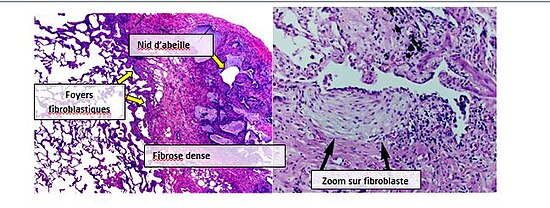
Même si un diagnostic pathologique de pneumopathie interstitielle usuelle correspond souvent à un diagnostic clinique de fibrose pulmonaire idiopathique, on peut observer un schéma histologique de pneumopathie interstitielle usuelle pour d’autres pathologies, notamment la fibrose d’origine connue (pathologies rhumatologiques par exemple). Il existe quatre caractéristiques principales de la pneumopathie interstitielle usuelle : fibrose interstitielle sur un modèle « patchwork », cicatrices interstitielles, nids d’abeilles et foyers fibroblastiques.
Les foyers fibroblastiques sont un ensemble dense de myofibroblastes et de tissus cicatriciel; avec les nids d’abeille, ce sont les principales découvertes pathologiques permettant de diagnostiquer la PIU.
Lavage broncho-alvéolaire
Le lavage broncho-alvéolaire est une procédure de diagnostic de la pneumopathie interstitielle qui est bien tolérée. L’examen cytologique du lavage broncho-alvéolaire (comptage des différentes cellules) doit être envisagé pour l’évaluation de patients atteints de fibrose pulmonaire idiopathique à discrétion du médecin traitant, selon la disponibilité et l’expérience de la structure de prise en charge. Le lavage broncho-alvéolaire peut mener à des diagnostics spécifiques différents : pathologies cancéreuses, infections, pneumonie à éosinophile, histiocytose langerhansienne, ou protéinose alvéolaire. Dans l’évaluation de patients avec suspicion de FPI, le lavage broncho-alvéolaire est principalement utilisé pour exclure tout autre diagnostic. Une lymphocytose marquée (>30 %) suggère généralement un diagnostic de FPI.
Tests de la fonction pulmonaire
Une spirométrie révèle habituellement une réduction de la capacité vitale soit avec réduction proportionnelle des volumes d’air, soit avec des volumes augmentés pour la capacité vitale observée. Cette dernière découverte reflète un manque de souplesse du poumon (compliance pulmonaire réduite) associé à une fibrose pulmonaire, ce qui provoque une diminution de l’élasticité du poumon.
La mesure des volumes pulmonaires statiques par pléthysmographie ou d’autres techniques révèle généralement des volumes pulmonaires réduits (restriction), ce qui reflète la difficulté à gonfler les poumons fibrotiques.
Les résultats du test de diffusion de l'oxyde de carbone(DLCO) sont toujours inférieurs en cas de fibrose pulmonaire idiopathique et peuvent représenter la seule anomalie en cas de fibrose pulmonaire idiopathique débutante ou légère. Cela montre la propension des patients atteints de fibrose pulmonaire idiopathique à une désaturation en oxygène à l’effort, ce qui peut aussi être évalué par le test de marche de 6 minutes (6MWT).
Les termes « légère », « modérée » et « sévère » sont parfois utilisés pour parler du stade de la maladie et dépendent souvent des résultats des tests de la fonction pulmonaire. Cependant il n’existe pas de consensus clair quant à la classification des stades de la maladie ou aux critères et valeurs de référence à utiliser. Une fibrose pulmonaire idiopathique légère à modérée a été décrite selon les critères fonctionnels suivants :
- capacité vitale forcée (CVF) : ≥ 50 % ;
- DLCO : ≥ 30 % ;
- distance 6MWT : ≥ 150 mètres.
Étude génétique en présence de FPI familiale
Autour de 10 et 15 % des patients atteints d'une FPI présentent une forme de fibrose pulmonaire retrouvée chez des membres de la même famille ; on parle alors de fibrose pulmonaire familiale. Il existe des mutations génétiques associées aux fibroses pulmonaires familiales (voir ci-dessus) et les tests de dépistage de ces mutations sont désormais employés en pratique médicale.
L'étude génétique constitue un volet de la démarche visant à apporter aux patients et à leur famille des informations sur la nature, la transmission et les conséquences de tels troubles génétiques. Il s'agit, en effet, de les aider à prendre des décisions éclairées d'ordre médical et personnel et aussi de connaître leur risque de voir se développer cette maladie héréditaire. Dans les cas où plus d'un membre de la famille est atteint de fibrose pulmonaire, il convient de proposer une étude génétique et un dépistage des mutations connues. L'étude génétique qui fait suite aux tests permet de personnaliser l'interprétation des résultats, pour savoir comment la santé du patient pourra être affectée tout comme son impact chez les autres membres de la famille.
Pronostic

L’évolution clinique de la fibrose pulmonaire idiopathique est médiocre La progression de la fibrose pulmonaire idiopathique est associée à une durée médiane de survie de 2 à 5 ans après diagnostic.
La survie à 5 ans pour la fibrose pulmonaire idiopathique représente entre 20 et 40 %, soit un taux de mortalité supérieur à celui de nombreuses pathologies cancéreuses, notamment celle du cancer du côlon, du myélome multiple et du cancer de la vessie.
Un indice multidimensionnel et un système d’évaluation des stades de la pathologie a été proposé pour prévoir la mortalité en cas de fibrose pulmonaire idiopathique. Cet indice s’appelle GAP et est basé sur le genre [G], l’âge [A], et deux variables pulmonaires physiologiques [P] [CVF et DLCO] généralement évaluées dans la pratique clinique pour évaluer la mortalité en cas de fibrose pulmonaire idiopathique. On a découvert que le dernier stade du GAP (stade III) est associé à un risque de mortalité de 39 % à un an. Ce modèle a également été testé dans les FPI et dans d'autres PI, c'est un bon outil pour prédire du pronostic vital dans tous les principaux sous-groupes de PI. Un score GAP-FPI modifié qui s'applique aux sous-groupes de FPI, a été mis au point pour prédire le pronostic vital associé à la maladie. Chez les patients atteints de FPI, le taux de mortalité global à 5 ans est élevé, cependant chez les patients ayant un déficit respiratoire léger à modéré le taux annuel de mortalité toutes causes confondues est relativement bas. C'est la raison pour laquelle la modification de la fonction respiratoire (CVF) mesurée à un an lors des essais cliniques est préférée au taux de survie.
En plus des paramètres cliniques et physiologiques utilisés pour prédire le devenir proche des patients atteints de FPI, les caractéristiques génétiques et moléculaires sont ajoutées pour évaluer la mortalité liée à la FPI. C'est ainsi qu'il est apparu que les patients atteints de FPI ayant un génotype spécifique au niveau du polymorphisme sur le gène promoteur de la mucine pulmonaire MUC5B (voir ci-dessus) ont une dégradation de leur FVC ralentie et un taux de survie significativement amélioré. De telles données sont certes intéressantes d'un point de vue scientifique, toutefois leur emploi dans la pratique clinique en tant que modèle pronostic pour des génotypes particuliers n'est pas encore à l'ordre du jour.
La fibrose pulmonaire idiopathique peut se compliquer d'une maladie thromboembolique, d'un cancer du poumon ou d'une hypertension artérielle pulmonaire.
Prise en charge
L’objectif du traitement de la fibrose pulmonaire idiopathique consiste essentiellement à faire diminuer les symptômes, enrayer la progression de la maladie, éviter les exacerbations aigues, et prolonger la survie. Les soins préventifs (ex. : vaccination) et le traitement en fonction des symptômes doivent être mis en œuvre chez tous les patients.
Interventions pharmacologiques
Par le passé, un certain nombre de traitements contre la fibrose pulmonaire idiopathique ont fait l’objet d’études, notamment ceux à base d’interféron gamma-1β, bosentan, ambrisentan, et d’anticoagulants, mais ils ne sont plus considérés comme des traitements efficaces. La plupart de ces études était basée sur l’hypothèse selon laquelle la fibrose pulmonaire idiopathique serait une maladie inflammatoire.
Pirfénidone
La pirfénidone est une petite molécule qui associe des effets anti-inflammatoires, antioxydants, et anti-fibrotiques sur des modèles expérimentaux de fibrose. La pirfénidone, commercialisée sous le nom commercial Esbriet, est légalement utilisée en Europe pour le traitement de patients atteints de fibrose pulmonaire idiopathique légère à modérée. Est elle aussi légale au Japon et en Corée du sud (nom commercial Pirespa), ainsi qu'au Canada, en Chine, en Inde, en Argentine et au Mexique.
La pirfénidone a reçu le feu vert de l’Union Européenne à la suite des résultats de trois études randomisées et contrôlées, en double aveugle et contre placebo, de phase III ; l’une a été réalisée au Japon, les deux autres en Europe et aux États-Unis (essais capacity).
Le risque de progression de la maladie chez les patients traités par pirfénidone diminue de 30 %. La CVF et la capacité vitale sont aussi nettement améliorées par la pirfenidone. À partir de ces résultats mitigés, la Food and Drug Administration. La pirfénidone a permis de réduire significativement la dégradation des fonctions respiratoires et de ralentir la progression de la FPI. Les données de l'étude ascend qui ont également été combinées avec les données provenant de deux études capacity dans une analyse prédéfinie ont montré que sous pirfénidone le risque de décès était abaissé de près de 50 % à un an. Sur la base de ces résultats, la pirfénidone a été reconnue comme médicament « Breakthrough Therapy Designation » par la FDA. Ce label est décerné par la FDA aux traitements qui visent à traiter des maladies ou des pathologies mortelles et lorsque les premiers résultats cliniques apportent la preuve que le produit est substantiellement plus efficace que les thérapies existantes, pour au moins un critère cliniquement significatif.
Le , l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) publie une mise à jour importante de sécurité et de nouvelles recommandations concernant les risques d’atteintes hépatiques d’origine médicamenteuse avec Esbriet (pirfénidone). Cet article précise que des cas graves de lésions hépatiques d’origine médicamenteuse ont été récemment signalés avec la pirfénidone, dont certains d'issue fatale. L'Agence recommande un bilan hépatique (ALAT, ASAT, bilirubine) avant l’initiation d’un traitement par pirfénidone. Par la suite, un bilan doit être réalisé mensuellement pendant les 6 premiers mois de traitement puis tous les 3 mois pendant toute la durée du traitement.
Nintédanib
Le nintedanib, inhibiteur triple de l'angiokinase et administré par voie orale, cible les récepteurs à activité tyrosine-kinase impliqués dans la régulation de l'angiogenèse : récepteur du facteur de croissance des fibroblastes (FGF), récepteur du facteur de croissance dérivé des plaquettes (PDGF) et le récepteur du facteur de croissance de l’endothélium vasculaire (VEGF-R), qui sont également impliqués dans la pathogenèse de la fibrose et de la FPI. Les deux essais cliniques de phase III ont montré que Nintedanib diminue la dégradation de la fonction respiratoire d'environ 50 % à un an. Concernant les critères secondaires, l'essai INPULSIS-2 est le seul a montré un allongement significatif du délai de survenue (apparition retardée) de la première exacerbation aigüe (voir ci-dessus) dans le groupe Nintedanib par rapport au groupe placebo. Cet allongement n'était pas visible dans l'essai INPULSIS-1.
Il n'existe pas d'étude comparant directement le nintedanib et la pirfénidone mais leurs résultats sont jugés comparables.
Antiacide
Les antiacides pourrait freiner l'évolution de la maladie, du moins sur des paramètres spirométriques. Cela n'est pas retrouvé dans toutes les études, d'autres trouvant une augmentation du risque infectieux.
N-acétylcystéine et trithérapie
La N-acétylcystéine est un précurseur du glutathion, un antioxydant. On a supposé que le traitement par doses élevées de N-acétylcystéine pourrait rétablir un déséquilibre oxydant/antioxydant existant dans les tissus pulmonaires du patient atteint de fibrose pulmonaire idiopathique. Lors du premier essai clinique sur 180 patients (ifigenia), la N-acétylcystéine a enrayé la chûte de capacité vitale et de DLCO sur 12 mois de suivi lorsqu’il était utilisé avec la prednisone et l'azathioprine (trithérapie).
Plus récemment, une grande étude randomisée contrôlée (panther-fpi) a été entreprise par le National Institutes of Health (NIH) aux États-Unis, afin d’évaluer la trithérapie et la monothérapie par N-acétylcystéine sur les patients atteints de fibrose pulmonaire idiopathique. Cette étude a démontré que l’association de prednisone, azathioprine, et N-acétylcystéine, augmentait le risque de décès et d’hospitalisations et le NIH a annoncé en 2012 que la partie de l’étude panther-fpi portant sur la trithérapie avait été interrompue de façon précoce. En conclusion, l'étude indique que « comparé au placebo, l'acétylcystéine n'apporte pas de bénéfice significatif quant au maintien de la CVF chez les patients atteints de fibrose pulmonaire idiopathique avec altération de la fonction respiratoire légère à modérée ». L'étude a également évalué la NAC administrée en monothérapie, les résultats obtenus pour ce bras ont été publiés récemment dans le New England Journal of Medecine, toutefois il apparait que la NAC prescrite en monothérapie n'apporte pas de bénéfices significatifs aux patients atteints de FPI légère à modérée.
Futures solutions thérapeutiques
Un certain nombre de substances sont en cours d'investigation dans le cadre d'essais cliniques de phase II. Il s'agit notamment d'anticorps monoclonaux tels que le simtuzumab (en), le tralokinumab (en), le lebrikizumab (en), le pamrevlumab, le FG-3019, un antagoniste du récepteur de l'acide lysophosphatidique (BMS-986020) et la pentraxine 2 recombinante. Ces molécules ciblent plusieurs cytokines et facteurs de croissance dont le rôle est connu dans la prolifération, l'activation, la différenciation ou la néfaste survie de fibroblastes.
Interventions non pharmacologiques
Greffe du poumon
La greffe du poumon peut être adaptée aux patients assez résistants pour être soumis à une opération chirurgicale importante. Chez les patients atteints de fibrose pulmonaire idiopathique, il a été démontré que la greffe du poumon réduit le risque de mortalité de 75 %, par rapport aux patients cantonnés à la liste d’attente. Depuis la création du Lung Allocation Score, qui détermine quels patients sont prioritaires pour recevoir une greffe en fonction de la survie estimée, la fibrose pulmonaire idiopathique est devenue la principale indication pour la greffe de poumon aux États-Unis.
Pour les patients symptomatiques atteints de fibrose pulmonaire idiopathique de moins de 65 ans et dont l’indice de masse corporelle est inférieur ou égal à 26 kg/m2 il faut envisager une greffe du poumon, mais il n’y a pas de données fiables quant à des délais précis. Même si cette opération est sujette à controverse, les données les plus récentes suggèrent que la greffe bilatérale donne de meilleurs résultats que la greffe unilatérale chez les patients atteints de fibrose pulmonaire idiopathique. On estime que la survie à 5 ans, après greffe du poumon chez les patients atteints de fibrose pulmonaire idiopathique, atteint entre 50 et 56 %.
Oxygénothérapie continue
L’oxygénothérapie, c’est-à-dire l’apport supplémentaire d’oxygène à domicile, est fortement conseillée dans les recommandations pour la fibrose pulmonaire idiopathique de 2011, pour les patients avec hypoxémie de repos cliniquement significative. Même s’il n’a pas été démontré que l’oxygénothérapie améliore la survie en cas de fibrose pulmonaire idiopathique, certaines données démontrent une amélioration de la capacité physique.
Réhabilitation respiratoire
La fatigue et la perte de masse musculaire sont courantes et handicapantes pour les patients atteints de fibrose pulmonaire idiopathique. La réhabilitation respiratoire peut alléger ou supprimer les symptômes de la fibrose pulmonaire idiopathique et améliorer le statut fonctionnel en stabilisant et/ou inversant les caractéristiques extra-pulmonaires de la maladie. Peu d'études ont été publiées sur le rôle de la rééducation respiratoire dans la prise en charge de la fibrose pulmonaire idiopathique, toutefois la plupart des études concluent à une amélioration significative à court terme de la capacité fonctionnelle respiratoire, de la qualité de vie et de la dyspnée à l'effort. Les programmes habituels de réhabilitation incluent des exercices physiques, de la modulation nutritionnelle, de la thérapie occupationnelle, de la formation et du conseil psychologique.Au cours de la dernière phase de la maladie, les patients atteints de fibrose pulmonaire idiopathique ont tendance à négliger l’activité physique en raison de la progression de la dyspnée. Lorsque cela est possible, il faut au contraire les encourager à poursuivre leurs activités physiques.
Soins palliatifs
L’objectif des soins palliatifs est de réduire les symptômes et d’améliorer le confort des patients plus que de traiter la maladie. Ils peuvent traiter les symptômes qui s’aggravent par l’utilisation d’opioïdes pour la dyspnée sévère et la toux. De plus, l’oxygénothérapie peut être utile pour pallier la dyspnée chez les patients hypoxémiques.
L’objectif des soins palliatifs est de soulager le patient sur le plan physique et émotionnel et d’apporter un soutien psychologique et psychosocial aux patients et à ceux qui leur prodiguent leurs soins. Au fur et à mesure que la maladie progresse, les patients peuvent ressentir une angoisse, de la peur, et même une dépression ; le soutien psychologique doit donc être envisagé. Dans une étude récente sur des patients en hôpital de jour atteints de pneumopathies interstitielles, notamment des fibroses pulmonaires idiopathiques, la dépression, le statut fonctionnel (évalué par le test de la marche), ainsi que la dégradation de la fonction pulmonaire ont contribué à la gravité de la dyspnée.
Dans certains cas de dyspnée particulièrement sévère, la morphine peut être utilisée. Elle peut réduire la dyspnée, l’angoisse et la toux sans diminution significative de la saturation en oxygène.
Suivi
La fibrose pulmonaire idiopathique est souvent mal diagnostiquée, du moins tant que les données physiologiques et émanant de l’imagerie suggèrent une pneumonie interstitielle, ce qui retarde le diagnostic et le traitement approprié. Étant donné qu’en cas de fibrose pulmonaire idiopathique la survie médiane après diagnostic est de trois ans, le renvoi précoce du patient vers une structure spécialisée doit être envisagé pour tout patient atteint de pneumonie interstitielle reconnue ou suspectée. Étant donné la complexité du diagnostic différentiel, il est capital, pour parvenir au bon diagnostic, de passer par un débat multidisciplinaire entre pneumologues, radiologues, et pathologistes expérimenté dans le diagnostic de la pneumopathie interstitielle.
Une fois la fibrose pulmonaire idiopathique diagnostiquée et le traitement choisi en fonction des symptômes et du stade de la maladie, un suivi étroit doit être mis en place. En raison de l’évolution variable de la maladie, de l’incidence élevée de complications comme un cancer du poumon (qui représente jusqu’à 25 % des patients atteints de fibrose pulmonaire idiopathique) un suivi de routine tous les trois à six mois est obligatoire, avec une spirométrie (pléthysmographie du corps entier), test de diffusion de l'oxyde de carbone, radio-thorax, 6MWT, évaluation de la dyspnée, de la qualité de vie, et du besoin en oxygène.
De plus, étant donné la prise de conscience des complications de la maladie et les pathologies fréquemment associées, une évaluation de routine des comorbidités est nécessaire (même si la plupart d’entre elles ne sont que le reflet du vieillissement) ainsi que du traitement, de ses interactions et effets indésirables.
Exacerbation aiguë
L’exacerbation aigüe consiste en une aggravation inexpliquée ou une progression de la dyspnée sur 30 jours, avec de nouvelles anomalies apparaissant à la tomodensitométrie, qui viennent s’ajouter à des images de pneumopathie interstitielle usuelle préexistante.
L’incidence annuelle de l’exacerbation aiguë représente entre 10 et 15 % de la totalité des patients. Le pronostic est mauvais, avec une mortalité entre 78 et 96 %. D’autres causes d’exacerbation comme l’embolie pulmonaire, l’insuffisance cardiaque congestive, le pneumothorax ou l’infection doivent être exclues. L’infection pulmonaire doit être exclue par aspiration endotrachéale ou lavage broncho-alvéolaire.
De nombreux patients subissant des exacerbations aiguës nécessitent un traitement intensif, en particulier lorsque l’insuffisance respiratoire est associée à une instabilité hémodynamique, à des comorbidités significatives ou à une hypoxémie sévère. Cependant, durant l’hospitalisation, la mortalité reste élevée. Une ventilation mécanique peut être mise en place après avoir longuement pondéré le pronostic à long terme et si possible le souhait du patient. Cependant les recommandations actuelles vont à l’encontre de l’utilisation de la ventilation mécanique chez les patients avec insuffisance respiratoire secondaire à une fibrose pulmonaire idiopathique.
Chez d’autres espèces
La fibrose pulmonaire idiopathique a été détectée chez plusieurs races de chiens et chats ; c’est chez les Westie (West Highland white terriers) qu’elle a le mieux été caractérisée. On retrouve chez les animaux la plupart des signes cliniques connus chez l’Homme, notamment une intolérance progressive à l’effort, un rythme respiratoire augmenté et une éventuelle détresse respiratoire. Le pronostic est généralement mauvais.
Liens externes
- Fibrose pulmonaire idiopathique sur le site de l'Open Directory Project
- Communauté de la fibrose pulmonaire idiopathique
- Fondation de la fibrose pulmonaire (Pulmonary Fibrosis Foundation)
- Communauté AIR (AIR Community)
- AIMIP : Associazione Italiana Malattie Interstiziali o Rare del Polmone
- Registre européen de la fibrose pulmonaire idiopathique, la première base de données européenne contenant des données transversales de patients atteints de fibrose pulmonaire idiopathique, notamment des groupes de contrôle de patients atteints d’autres pathologies pulmonaires
- Fondation ILD Care Foundation dont les activités se concentrent sur l’amélioration des connaissances, le soutien à la recherche, la contribution à la prévention et l'établissement de conseils sur les pathologies interstitielles