

La neuro-oncologie (du grec ancien νεῦρον neuron, français «nerf», ὄγκος onkos, français «gonflement» et -λογία -logia, français « enseignement », « science ») est un domaine de la médecine qui combine la neurologie et l'oncologie.
La neuro-oncologie s'occupe principalement du diagnostic, de la thérapie et de la recherche des maladies tumorales du système nerveux, qui comprend les types de tumeurs suivants:
- tumeurs primitives du système nerveux central (tumeurs cérébrales et tumeurs de la moelle épinière);
- tumeurs secondaires du système nerveux central (métastases cérébrales et métastases médullaires);
- lymphomes malins du système nerveux central;
- tumeurs du système nerveux périphérique.
Cette liste montre déjà qu'il s'agit d'un vaste domaine difficile à cerner. Bien que le traitement des lymphocytomes du système nerveux central suive également les directives de traitement hématologique et que les métastases cérébrales ne soient pas traitées séparément du traitement de base de la tumeur primaire, les tumeurs des nerfs périphériques sont bénignes, comme les neurinomes, et comme les autres tumeurs des tissus mous, elles sont souvent localisées dans l'orthopédie région.
Informations générales
Tumeurs primitives du système nerveux central
Les tumeurs cérébrales primitives peuvent survenir à tout âge, de la petite enfance à la fin de la vie. Des facteurs tels que l'âge, l'emplacement de la tumeur et la présentation clinique sont utiles dans le diagnostic différentiel. La plupart des types de tumeurs cérébrales primaires sont plus fréquentes chez les hommes, à l'exception des méningiomes, qui sont plus fréquents chez les femmes.
Tumeurs métastatiques du système nerveux central
L'invasion ou la compression directe des tissus continus est liée à la proximité du système nerveux avec d'autres structures.
Métastases intracrâniennes
Il existe trois types de métastases intracrâniennes: les métastases cérébrales, les métastases durales et les métastases méningées-leptoméningées. Les métastases cérébrales peuvent être uniques ou multiples et affecter n'importe quelle partie du cerveau. Les métastases aux structures durales se produisent généralement par propagation hématogène ou invasion directe à partir d'un os adjacent. Les métastases durales peuvent envahir le cerveau sous-jacent et provoquer un œdème focal et des symptômes neurologiques associés. En raison de leur localisation corticale, ces processus ont tendance à provoquer des crises au début du cours. La métastase leptoméningée est un phénomène clinique rare mais bien connu chez les patients cancéreux. Les métastases leptoméningées sont le plus souvent dues à des tumeurs primitives du sein, du poumon ou d'un mélanome.
Métastases crâniennes
Les métastases crâniennes sont divisées en deux catégories: Calvarium et base du crâne.
Tumeurs primitives du système nerveux central
Une première subdivision des tumeurs du système nerveux central (SNC) se situe entre les tumeurs primaires (provenant directement du SNC) et métastatiques (provenant d'un autre organe). Ces derniers ont une incidence environ dix fois supérieure à celle des premiers. Les tumeurs cérébrales sont des néoplasmes qui se développent dans le cerveau. Les tumeurs telles que le méningiome, qui, en raison de leur masse comprimant mais ne pénétrant pas le cerveau, et les tumeurs hypophysaires et épiphysaires, qui sont situées sur le tronc cérébral, sont souvent appelées à tort tumeurs cérébrales. Le terme de tumeurs intracrâniennes les résume plus précisément.
Les tumeurs primaires du SNC comprennent une variété d'entités pathologiques, chacune avec sa propre histoire naturelle. Du fait que les tumeurs gliales représentent à elles seules près de 40 % de ces tumeurs, on peut d'abord distinguer les tumeurs gliales (gliomes) des tumeurs non gliales. Les gliomes les plus courants sont les astrocytomes (provenant des cellules astrocytaires gliales), les oligodendrogliomes (provenant des cellules oligodendrogliales) et les épendymomes (provenant des cellules épendymaires).
Épidémiologie
Les tumeurs primitives malignes du système nerveux central sont relativement rares et représentent environ 2 % de toutes les tumeurs malignes. Les maladies tumorales du système nerveux central sont distribuées à 95 % sur le cerveau et à 5 % sur les méninges, les nerfs crâniens et la moelle épinière. Ils peuvent survenir à tout âge et le risque de développer la maladie augmente avec l'âge. Chez l'adulte, les gliomes peuvent être retrouvés histologiquement à partir du tissu de soutien des cellules nerveuses, dont environ 75 % de glioblastomes Astrocytomes IV. degrés avec un pronostic défavorable. Les tumeurs embryonnaires prédominent chez les nourrissons et les jeunes enfants. En Allemagne, environ 3 970 personnes sont tombées malades en 2 016 hommes et 3 460 femmes atteintes de tumeurs malignes du système nerveux central. En moyenne, les taux de survie sont de 21 % pour les hommes et 24 % pour les femmes. Les statistiques incluent également rarement les tumeurs histologiquement bénignes du SNC, qui surviennent dans environ 6 000 nouveaux cas par mensonge de l'année. Environ 65 % provient des méninges. Les femmes sont beaucoup plus touchées. Si elles ne sont pas traitées par chirurgie ou radiothérapie, même les tumeurs bénignes peuvent être mortelles en raison de la croissance progressive dans l'espace crânien fermé. La tumeur maligne intracrânienne du SNC la plus fréquente est le glioblastome, la plus bénigne étant le méningiome.
Étiologie
La prédisposition génétique aux néoplasmes du système nerveux central est relativement rare, bien que certains gliomes puissent survenir en tant que complications de plusieurs maladies familiales.
La mutation de certains gènes suppresseurs de tumeurs caractérise plusieurs syndromes héréditaires qui montrent une susceptibilité accrue au développement de tumeurs cérébrales. Les mutations suivantes et leurs syndromes sont associés à un risque plus élevé de développer des tumeurs cérébrales : mutation du gène NF1 avec neurofibromatose de type 1, mutation APC avec syndrome de Turcot, mutation PTCH avec syndrome de Gorlin et TP53 ou CHEK2 -Mutation avec syndrome de Li-Fraumeni.
Les facteurs environnementaux associés aux tumeurs cérébrales primitives sont difficiles à identifier. Dans certaines études, l'exposition au chlorure de vinyle a été associée à une incidence accrue de gliomes de haut grade. La seule cause rare mais bien identifiée d'une tumeur cérébrale primitive est le rayonnement ionisant. En particulier, la radiothérapie des enfants atteints de teigne du cuir chevelu et des patients atteints de leucémie lymphatique aiguë, de craniopharyngiome ou de lymphome non hodgkinien est associée à un risque accru de gliome. Il existe un risque accru de lymphome cérébral primitif chez les patients atteints du SIDA.
Clinique
Signes et symptômes
Les symptômes de la néoplasie cérébrale sont caractérisés par un déplacement ou destruction des tissus environnants et infiltration des mêmes causes.
Le symptôme le plus courant, signalé par 35 % des patients, est le mal de tête. La survenue de céphalées sévères chez des patients qui en souffrent rarement est souvent caractéristique, surtout si les crises de céphalées ou les migraines sont plus sévères le matin et s'accompagnent de nausées, de vomissements et de déficits neurologiques. Chez les patients qui souffrent plus souvent de maux de tête, un changement de forme, une augmentation de la fréquence ou de l'intensité des crises peuvent être un symptôme du développement d'une tumeur cérébrale. Des convulsions surviennent chez environ un tiers des patients atteints de gliome, en particulier avec des tumeurs de bas grade ou du SNC. Les déficits neurologiques focaux sont liés à la localisation de la tumeur. Des modifications de l'état mental se produisent également chez 15 à 20 % des patients atteints de gliome.
Diagnostic d'imagerie

La tomodensitométrie (TDM) et L'imagerie par résonance magnétique (IRM) peuvent détecter efficacement une néoplasie dans le cerveau. L'IRM est plus sensible que la TDM pour identifier les lésions, mais présente des contre- indications pour les patients porteurs de stimulateurs cardiaques, de prothèses incompatibles, de clips métalliques et contre-indications. La TDM reste la méthode de choix pour détecter les calcifications au sein des lésions ou les érosions osseuses de la calotte ou de base du crâne. L'utilisation d' agents de contraste, iodés dans le cas du scanner et paramagnétiques (gadolinium) dans le cas de l'IRM, permet l'acquisition d'informations sur la vascularisation et l'intégrité de la barrière hémato-encéphalique, une meilleure définition de la tumeur tumorale par rapport à l' œdème environnant et à la génération d' hypothèses sur le degré de malignité. L'examen radiologique permet également d'évaluer les effets mécaniques et les modifications conséquentes des structures cérébrales résultant de la tumeur, telles que l' hydrocéphalie et les hernies, dont les effets peuvent être fatals. Enfin, en préparation à la chirurgie, ce diagnostic peut être utilisé pour déterminer la localisation de la lésion ou l'infiltration de la tumeur dans des zones vitales du cerveau. À cette fin, l'IRM est plus efficace que la tomodensitométrie car elle peut fournir des images en trois dimensions.

Les outils d'imagerie radiologique diagnostique mettent en évidence la modification du tissu néoplasique par rapport au parenchyme cérébral normal (par le biais de modifications de la densité tissulaire imagée électroniquement en TDM et de l'intensité du signal en IRM). Comme la plupart des tissus pathologiques, les tumeurs sont également reconnaissables par une accumulation accrue d'eau intracellulaire. Dans la tomodensitométrie, ils apparaissent hypodenses, c'est-à-dire de moindre densité que le parenchyme cérébral, dans la tomographie par résonance magnétique nucléaire avec relaxation spin-réseau hypointense et en relaxation spin-spin ainsi que l' hypersignal en pondération protonique (PD).
La zone saine du cerveau ne doit présenter aucune luminescence particulière sur une image radiologique. Par conséquent, il va sans dire que l'attention est portée sur des plages de signaux de contraste plus larges.
Dans le tissu tumoral, en général, la plus grande proportion d'amélioration du contraste est due à la barrière hémato-tumorale particulière qui permet le passage de l'iode (CT) et du gadolinium (IRM) dans l'espace interstitiel extravasculaire intratumoral. Cela augmente le signal (densité ou intensité) de la tumeur. Cependant, des précautions doivent être prises pour s'assurer que l'amélioration du contraste ne différencie pas définitivement la néoplasie de l'œdème péri-lésionnel. En fait, la découverte anatomo-pathologique dans le tissu tumoral infiltrant malin du gliome, comme dans le glioblastome et l'astrocytome anaplasique, montre également au-delà de l'œdème vasogénique causé par la destruction de la barrière hémato-encéphalique par la tumeur. Cette dernière condition clinique est difficilement détectable par imagerie diagnostique.
La tomodensitométrie du cerveau montre généralement une masse tissulaire qui peut être améliorée par l'un ou l'autre contraste. Au scanner, les gliomes de bas grade apparaissent généralement isodenses au parenchyme normal et peuvent donc ne pas présenter de rehaussement de contraste. De même, les lésions de la fosse crânienne postérieure sont difficiles à identifier au scanner. Par conséquent, les seuls résultats d'une telle tomographie ne sont pas toujours suffisants à des fins diagnostiques. Dans les cas douteux, l'utilisation de l'imagerie par résonance magnétique plus sensible est indispensable.
Sur-L'IRM montre une tumeur intracrânienne comme une lésion massive qui peut devenir plus luminescente après utilisation du produit de contraste. Cependant, il y a toujours une anomalie de signal dans -L'imagerie par résonance magnétique, qui indique la présence d'une néoplasie ou d'un œdème vasogénique. Habituellement, une luminescence accrue (amélioration du contraste) indique une tumeur d'un grade supérieur de malignité. Un anneau de contraste est caractéristique du glioblastome, avec la partie luminescente correspondant à la partie vitale de la tumeur maligne, et la plus foncée - zone hypointense correspondant à une nécrose tissulaire.
Mise en scène
La plupart des tumeurs intracrâniennes primaires restent localisées dans le crâne, de sorte que les procédures de stadification systémique ne sont pas nécessaires.
En revanche, les tumeurs neuroectodermiques primaires, les médulloblastomes, les tumeurs germinales du SNC et les lymphomes primaires du SNC se propagent souvent via l'espace sous - arachnoïdien jusqu'aux leptoméninges. Une imagerie par résonance magnétique rachidienne ou une ponction lombaire est donc également nécessaire pour tous les patients présentant de tels diagnostics.
Types de tumeurs
Gliomes
Les tumeurs primaires du système nerveux central (SNC) impliquent une variété de tissus pathologiques, chacun avec sa propre histoire naturelle. En raison du fait que les gliomes représentent à eux seuls près de 40 % de toutes les tumeurs du SNC, il est courant dans la littérature de faire la distinction entre les tumeurs gliales et non gliales.
Astrocytome
Divers systèmes de catégories ont été proposés dans la littérature au fil du temps pour classer la malignité des astrocytomes. Depuis 1993, le système d'évaluation à quatre niveaux proposé par l' Organisation mondiale de la santé (OMS) est le plus largement utilisé et appliqué. Elle est basée sur quatre caractéristiques histologiques : augmentation de la densité cellulaire, mitose, prolifération endothéliale et nécrose. Par la suite, les astrocytomes de grade I, tels que les astrocytomes pilocytiques, sont généralement d'histologie bénigne. Astrocytomes II. Les grades (diffus) montrent une densité cellulaire accrue comme seule caractéristique histologique et sont des néoplasmes avec un degré d'infiltration inférieur. Les astrocytomes III montrent une mitose importante. grade (anaplasique). Et la prolifération ou la nécrose endothéliale sont observées dans les astrocytomes IV. degrés, les soi-disant glioblastomes.
Astrocytomes de bas grade



Les astrocytomes pilocytiques (y compris l'astrocytome pilomyxoïde), les astrocytomes sous-épendymaires à cellules géantes et les xanthastrocytomes pléomorphes font partie des tumeurs qui ont été décrites. Ce sont des néoplasmes un peu plus rares d'histologie bénigne qui ne peuvent souvent être guéris que par chirurgie. Si l' excision est incomplète, le tissu tumoral restant pourrait être traité avec succès par radiothérapie. Dans les rares cas où le traitement local ne fonctionne pas, la chimiothérapie systémique peut réussir, ce qui doit être ajusté individuellement. Les enfants répondent à une combinaison de carboplatine et de vincristine.
Les astrocytomes diffus II apparaissent sur la tomodensitométrie. Grades que les lésions moins intenses. Dans l'imagerie par résonance magnétique préférée, les agents de contraste peuvent ne pas être en mesure de mettre en évidence ces néoplasmes, leur luminescence peut être plus fine et plus faible. Un plus intense peut indiquer des tissus d'anaplasie accrue. Dans la mesure du possible, une biopsie est suggérée pour obtenir des échantillons de la partie anaplasique de la tumeur.
Dans la plupart des cas, les patients atteints d'astrocytomes diffus sont âgés de 20 à 40 ans. La survenue de crises d'épilepsie est typique pour eux. Les conditions d'un pronostic favorable sont le jeune âge, la taille de la tumeur inférieure à 50 millimètres et la résection chirurgicale la plus étendue possible de la tumeur. Les récidives tardives sont relativement fréquentes, c'est pourquoi les patients doivent être suivis pendant 15 ans après l'ablation de la tumeur.
Malgré leur évolution relativement lente, la plupart des astrocytomes évoluent vers des lésions caractérisées par une anaplasie étendue qui sont généralement réfractaires à la chirurgie et à la radiothérapie. Cependant, le traitement des patients atteints d'astrocytomes diffus de bas grade ne fait pas l'unanimité dans la littérature. Le rôle de la résection complète est discuté dans des contextes professionnels. Les résultats de certaines études montrent que l'ablation maximale de la tumeur donne les meilleurs résultats. En fait, les petites tumeurs unilatérales peuvent être complètement éliminées si aucune structure critique du cerveau n'est impliquée. Une approche pragmatique généralement acceptable pour la généralité des cas consiste à retirer la néoplasie dans la mesure du possible afin d'éviter des déficits neurologiques importants.
Des études ont montré que la radiothérapie administrée immédiatement après le diagnostic a prolongé la période pendant laquelle le patient est sans maladie avant la récidive de la tumeur par rapport à la situation où le cours de la radiothérapie est retardé jusqu'au moment de la progression. Cependant, il n'existe actuellement aucun consensus sur le fait que la radiothérapie peu de temps après le diagnostic améliore la « survie globale » du patient.
Chez les patients présentant des symptômes plus légers ou inexistants, ou des crises pouvant être contrôlées par des anticonvulsivants, il est possible de retarder la radiothérapie jusqu'à ce que la croissance tumorale atteigne une phase critique. On souhaite souvent réduire le risque de dommages neurologiques causés par la radiothérapie elle-même.
Deux essais cliniques randomisés prospectifs n'ont montré aucun bénéfice supérieur de la radiothérapie à haute dose par rapport à la radiothérapie à faible dose. Typiquement, le dosage total est compris entre 45 et 54 Gray avec un fractionnement de 1,8 à 2 Gray.
L'effet de la chimiothérapie adjuvante chez les patients atteints d'astrocytomes de bas grade est toujours à l'étude. Les résultats préliminaires d'un essai clinique comparant la radiothérapie seule à la radiothérapie suivie d'une chimiothérapie contenant de la procarbazine, de la lomustine et de la vincristine (PCV) ont montré une plus longue période de "survie sans maladie" avec l'association, mais pas de "survie globale" prolongée. En raison de la toxicité associée au protocole PCV, l'utilisation du témozolomide est recommandée à la fois comme traitement initial et après la guérison.
Astrocytomes anaplasiques
L'astrocytome anaplasique est une tumeur cérébrale maligne caractérisée par une croissance diffuse, une densité cellulaire accrue et des figures de division nucléaire. Elle est issue d'une population cellulaire spécifique du système nerveux central, les astrocytes. Selon la classification OMS des tumeurs du système nerveux central, la tumeur correspond à une tumeur de grade III.
En règle générale, les patients atteints d'astrocytome anaplasique présentent des crises d'épilepsie, des déficits neurologiques focaux, des maux de tête et des changements de personnalité. L'âge moyen des patients est de 45 ans. L'imagerie par résonance magnétique montre généralement une lésion massive avec un signal de contraste accru, qui peut aussi être plus faible. Le diagnostic repose sur l'examen histologique de la lésion par biopsie ou résection chirurgicale.
Un pronostic plus sombre peut être associé à un âge avancé, à une mauvaise condition physique et à des dommages neurologiques importants. En général, le résultat thérapeutique est meilleur avec une exérèse chirurgicale complète (traitement standard) sans augmentation des déficits neurologiques. La radiothérapie est la norme car il a été démontré qu'elle augmente le temps de survie. Le rôle de la chimiothérapie est controversé.
Glioblastome

Les tumeurs des cellules gliales les plus courantes et les plus malignes sont les glioblastomes. Ils consistent en une masse hétérogène de cellules d'astrocytome peu différenciées principalement chez l'adulte. Ils surviennent généralement dans les hémisphères cérébraux, plus rarement dans le tronc cérébral ou la moelle épinière. Sauf dans de très rares cas, comme toutes les tumeurs cérébrales, elles ne s'étendent pas au-delà des structures du système nerveux central.
Le glioblastome peut provenir d'une forme diffuse (II. grade) ou un astrocytome anaplasique (III. grade) développer. Dans ce dernier cas, il est dit secondaire. Cependant, lorsqu'elle survient sans antécédent ni signe de malignité antérieure, on parle de maladie primaire. Les glioblastomes sont traités par chirurgie, radiothérapie et chimiothérapie. Ils sont difficiles à guérir et rares sont les cas qui survivent au-delà de trois ans.
Oligodendrogliomes
Oligodendrogliome est une tumeur cérébrale gliale rare qui provient des oligodendrocytes. Elle survient principalement chez l'adulte entre 40 et 45 ans, préférentiellement dans le cortex cérébral et la substance blanche des hémisphères cérébraux.
Les oligodendrogliomes sont relativement rares, représentant moins d'environ 5 % de toutes les tumeurs cérébrales primaires et pas plus d'environ 10 à 15 % de tous les gliomes. Ces tumeurs sont divisées en lésions de bas grade et anaplasiques. L'oligodendrogliome anaplasique est caractérisé par une augmentation de la densité cellulaire, de la mitose, de la prolifération endothéliale et du polymorphisme nucléaire, et de la nécrose.
Oligodendrogliomes et oligoastrocytomes de bas grade
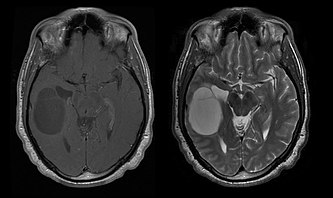


La survie médiane des patients atteints d' oligodendrogliome pur est d'environ 10 ans, avec un oligoastrocytome d'environ 8 ans. L'allongement par rapport aux astrocytomes purs est dû à une délétion ou une translocation de la paire 1p/19q dans la tumeur.
L'âge moyen des patients au moment du diagnostic est de 35 ans. Les symptômes typiques sont des crises d'épilepsie, mais des déficits neurologiques focaux, des changements de personnalité ou d'autres symptômes de pression intracrânienne, tels que des maux de tête et des vomissements, peuvent également être signalés. Ces tumeurs ne sont généralement pas visibles sur la tomodensitométrie, l' IRM est donc la méthode de choix pour l'imagerie diagnostique. Sur le - Imagerie par résonance magnétique, ils sont reconnaissables à une intensité de signal accrue. sur -images, d'autre part, le signal peut être mis en sourdine et l'amélioration du contraste ne peut être détectée qu'occasionnellement. Un signal de calcification peut manquer.
Ces tumeurs progressent plus lentement que les astrocytomes de bas grade et il n'y a pas de consensus dans la littérature concernant un traitement optimal. Le traitement initial consiste à contrôler les symptômes avec des anticonvulsivants, une radiothérapie, une chimiothérapie ou une combinaison des deux derniers. La chirurgie, la radiothérapie et la chimiothérapie jouent un rôle important dans les rechutes. Les résections peuvent soulager les symptômes. Le témozolomide a montré 50 % de patients qui rechutent après la radiothérapie ont une réaction positive.
Oligodendrogliomes anaplasiques
Les oligodendrogliomes anaplasiques présentent des symptômes typiques résultant de l'effet de masse et des crises d'épilepsie. Malgré leur chimiosensibilité, la médiane de survie n'est que de 3 jusqu'à 5 années. Le traitement consiste en une excision maximale, suivie d' une radiothérapie. Concernant la chimiothérapie, il est à noter que deux essais cliniques de phase III récents ont comparé les résultats de la radiothérapie à ceux de la radiothérapie combinée et de la chimiothérapie procarbazine, lomustine, vincristine. Bien que la survie sans symptômes pertinents ait été plus longue avec la thérapie combinée, la survie globale était la même pour les deux thérapies. Les patients avec une délétion 1p/19q ont obtenu les meilleurs résultats de traitement, tandis que les patients sans délétion 1p/19q ont pu améliorer leurs résultats avec la chimiothérapie PCV.
Des études cliniques prospectives ont montré qu'environ 50 jusqu'en 70 % de patients atteints d' oligodendrogliome anaplasique récurrent après radiothérapie qui répondent positivement à la chimiothérapie par PCV ou témozolomide. Bien que l'efficacité supérieure du témozolomide et du traitement PCV n'ait pas été établie, l'absence de myélosuppression cumulative avec le témozolomide suggère son utilisation au début du traitement des rechutes.
Épendymomes


L'épendymome est un néoplasme qui se développe à partir de cellules épendymaires tapissant les ventricules cérébraux, le plexus choroïde, le filum terminale et le canal central de la moelle épinière . Des cellules épendymaires sont également présentes dans le parenchyme cérébral à la suite de la migration embryonnaire des zones périventriculaires vers le cortex cérébral.
Ces tumeurs assez rares peuvent apparaître à tout âge, mais elles présentent deux pics caractéristiques, de 0 à 10 ans et de 40 à 50 ans. Les lésions intracrâniennes, qui surviennent généralement dans la fosse postérieure, sont plus fréquentes dans le premier groupe d'âge, tandis que les lésions de la colonne vertébrale sont plus fréquentes dans le deuxième groupe d'âge.
Les épendymomes sont divisés en lésions de bas grade (I. et II. grade sur l'échelle de l'OMS) et les lésions anaplasiques (III. degrés) subdivisé. I. grade sont en particulier les sous-épendymomes et les épendymomes myxopapillaires, III. Épendymome anaplasique. Les patients atteints d'épendymomes de bas grade dans la colonne vertébrale qui peuvent être complètement retirés ne subissent pas de radiothérapie par la suite. La place de la radiothérapie postopératoire dans les épendymomes intracrâniens de bas grade est controversée, mais le traitement par radiothérapie est généralement indiqué pour les tumeurs anaplasiques ou de bas grade qui ne peuvent pas être complètement réséquées.
Des études cliniques ont montré que les épendymomes répondent aux chimiothérapies, en particulier celles à base de platine . Le bénéfice de la chimiothérapie à base de platine est de 67 Pourcentage, en revanche 25 pour les nitrosourées Pour cent. Le pronostic des épendymomes II. Les grades sont la survie sans maladie à 6 ans de 68 pour cent et avec une survie globale de 87 Pour cent. Dans les épendymomes anaplasiques, ces valeurs chutent à 29 pour cent ou à 37 Pour cent.
Tumeurs non gliales
Medulloblastomes
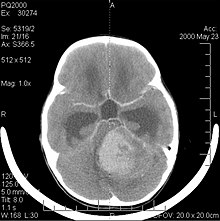
Le médulloblastome est la tumeur cérébrale maligne la plus fréquente chez l'enfant. L'incidence la plus élevée survient chez les enfants âgés de 2 à 7 ans. Le plus grand risque de maladie demeure dans l'enfance, car le médulloblastome est très rare chez les personnes de plus de 21 ans.
Cette tumeur est typique de la fosse postérieure, où elle est localisée dans les deux hémisphères du cervelet ou dans le vermis cérébelleux. Parce qu'il est envahissant et à croissance rapide, il se propage généralement à d'autres parties du système nerveux central (SNC) via le LCR et peut infiltrer le plancher du quatrième ventricule voisin et les méninges. Plus rarement, des métastases supplémentaires du SNC peuvent survenir. Lorsque la tumeur maligne survient, les symptômes comprennent la perte d'équilibre, l'incoordination, la diplopie, la dysarthrie et l'atteinte du quatrième ventricule, ce qui entraîne souvent une hydrocéphalie obstructive, des maux de tête, des nausées et des vomissements et une démarche instable.
L'IRM montre généralement une lésion de contraste massive impliquant le cervelet. Comme mentionné ci-dessus, le médulloblastome a une forte propension à infiltrer localement les leptoméninges ainsi qu'à se propager à travers l'espace sous-arachnoïdien, impliquant les ventricules, la convexité cérébrale et les surfaces leptoméningées de la colonne vertébrale. Par conséquent, il est nécessaire de mettre en résonance tout l'axe crânio-spinal.
Le but de la chirurgie est d'enlever autant que possible la masse présentée par la lésion. En effet, les tumeurs résiduelles postopératoires entraînent un moins bon pronostic. La présence de cellules tumorales dans le liquide céphalo-rachidien ou la détection par résonance de métastases leptoméningées est également un signe avant-coureur d'un pronostic défavorable. La chirurgie seule n'est généralement pas curative. Dans certains cas, cependant, une irradiation thérapeutique de l'axe craniospinal, focalisée sur le site tumoral primaire, peut en résulter. L'ajout d'une chimiothérapie après la radiothérapie augmente le taux de guérison. Des médicaments à base de platine (cisplatine ou carboplatine), de l'étoposide et un agent alkylant (cyclophosphamide ou lomustine) sont utilisés avec la vincristine. Avec un traitement approprié, les cas de longue survie de plus de 3 ans chez les patients atteints de médulloblastome varient de 60 à 60 ans et 80 Pour cent.
Méningiomes

Les méningiomes sont les tumeurs cérébrales extrinsèques ou extra-axiales intracrâniennes les plus courantes qui proviennent des cellules de l'arachnoïde, la membrane qui recouvre le cerveau et la moelle épinière. L'incidence de cette néoplasie est d'environ 2 cas par an pour 100 000 habitants. Ils sont plus fréquents chez les femmes dans leurs sixième et septième décennies. Leur fréquence est plus élevée chez les patients atteints de neurofibromatose de type 2. La perte du chromosome 22 est caractéristique des méningiomes, bien que la signification pronostique de cette découverte soit encore incertaine.
Les patients atteints de méningiome peuvent présenter des symptômes typiques d'une lésion crânienne massive, notamment des convulsions et des déficits neurologiques focaux. détecté sur la tomodensitométrie et l'imagerie par résonance magnétique pour d'autres raisons. Cette tumeur de résonance a un aspect caractéristique, consistant généralement en un rehaussement de contraste uniforme le long de la dure-mère avec une séparation nette du parenchyme cérébral. Une autre caractéristique, bien que non présente dans tous les cas, est la soi-disant "queue durale", représentée par un renflement qui s'étend au-delà de la lésion et indique le point d'ancrage dans la dure-mère.
De nombreux méningiomes découverts fortuitement ne nécessitent pas de traitement au moment du diagnostic initial. Si le patient présente un effet de masse significatif, que les symptômes soient présents ou non, le traitement de choix est généralement complet résection. Dans un studio Mayo Clinics comparant les taux de contrôle des tumeurs après résection chirurgicale et radiochirurgie chez des patients atteints de méningiome intracrânien petit à modéré et sans symptômes d'effet de masse, la radiochirurgie a permis un meilleur contrôle (98 contre 88 %) et avec moins de complications (10 contre 22 %) par rapport à l'ablation chirurgicale.
Lymphomes primitifs du SNC

Le lymphome primitif du système nerveux central représente environ 2 pour cent à 3 pour cent de toutes les tumeurs cérébrales chez les patients ayant un système immunitaire normal. Ils surviennent plus fréquemment chez les hommes de plus de 55 ans jusqu'à 60 ans. Près de la moitié de tous les lymphomes surviennent chez des patients de plus de 60 ans et environ un quart chez des patients de plus de 70 ans ans. L'incidence semble augmenter avec l'âge, mais la raison n'est pas encore claire. Les patients dont le système immunitaire est affaibli sont plus à risque de développer un lymphome du SNC, de sorte que ceux qui ont subi une greffe d'organe ont une immunodéficience congénitale ou une maladie auto-immune, ou sont infectés par le virus de l'immunodéficience humaine. Les lymphomes cérébraux associés au VIH sont associés au virus d'Epstein-Barr, en particulier chez les patients dont le nombre de lymphocytes CD4 est inférieur à 500 cellules par millimètre cube dans le sang. La plupart des lymphomes du SNC sont des lymphomes diffus à grandes cellules B.
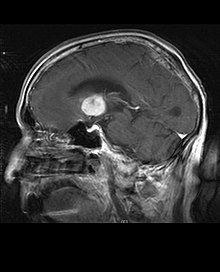
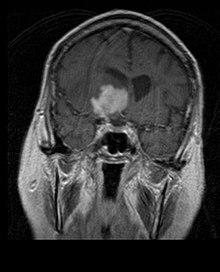
Les patients souffrent d'une variété de symptômes caractéristiques d'une lésion massive focale ou multifocale. L'IRM montre généralement des tumeurs avec un rehaussement de contraste homogène au sein de la substance blanche périventriculaire profonde. La multifocalité et le rehaussement inhomogène sont typiques des patients dont le système immunitaire est affaibli. L'analyse du lymphome du SNC est extrêmement importante dans le diagnostic différentiel de la néoplasie cérébrale. Il est à noter que l'administration de corticoïdes peut entraîner la disparition complète du rehaussement, rendant difficile le diagnostic des lésions. Par conséquent, si un lymphome du SNC doit être pris en compte dans le diagnostic différentiel, les corticoïdes doivent être évités à moins que l'effet de masse ne provoque un problème grave et immédiat chez le patient.
La biopsie de la lésion suspectée est cruciale. Contrairement au lymphome systémique à grandes cellules B, dans lequel la chimiothérapie et la radiothérapie sont efficaces et le traitement des lésions localisées est curatif, le lymphome du système nerveux central répond généralement au traitement initial mais réapparaît ensuite. Comme pour le lymphome systémique, le rôle de la chirurgie est principalement limité à l'obtention d'échantillons de tissus appropriés pour le diagnostic.
Dans le passé, la radiothérapie était administrée à l'ensemble du cerveau (panencéphalique). La médiane de survie est d'environ 12 même avec des lésions localisées Mois. La récidive affecte généralement le site de la blessure précédente ainsi que d'autres régions. Les réponses à la chimiothérapie sont plus prometteuses. Les essais cliniques dans lesquels le méthotrexate à haute dose seul a été utilisé comme premier traitement et la radiothérapie a été retardée jusqu'au moment de la rechute ou de la progression ont montré une meilleure survie globale que la radiothérapie seule. Encore plus efficace était la combinaison de méthotrexate, vincristine, procarbazine, méthotrexate intrathécal, cytarabine et radiothérapie panencéphalique et cytarabine, ou l'utilisation d'une chimiothérapie intra-artérielle avec méthotrexate intra-artériel, cyclophosphamide injecté par voie intraveineuse et étoposide après modification du sang. barrière cérébrale avec du mannitol. La médiane de survie sous méthotrexate était de 24 jusqu'à 40 mois beaucoup plus élevé qu'avec la radiothérapie seule (extrêmes 24 jusqu'à 40 mois). Dans certains cas, la radiothérapie n'est utilisée que pour les rechutes lorsqu'il y a une régression initiale avec la chimiothérapie. Des cas de survie longue ont également été rapportés sans radiothérapie.
La radiothérapie panencéphalique est associée à un risque élevé de développer une démence ou une leucoencéphalopathie . Ce risque pourrait être réduit en développant des stratégies efficaces de contrôle des tumeurs qui évitent la radiothérapie panencéphalique. Le traitement initial des patients dont le système immunitaire est affaibli consiste à réduire les causes de l'immunosuppression. Le pronostic de ces patients est généralement pire que celui des patients dont le système immunitaire est normal. En raison des infections tumorales qui l'accompagnent et d'une condition physique généralement sous-optimale, la chimiothérapie ne peut souvent pas être effectuée chez ces patients immunodéprimés. Comme pour les autres tumeurs cérébrales, la réponse aux traitements dépend de l'âge et de la condition physique.
Tumeurs métastatiques du système nerveux central
Métastases cérébrales
Les métastases cérébrales sont les néoplasmes intracrâniens les plus courants chez les adultes, étant dix fois plus fréquents que les tumeurs cérébrales primaires. Ils marchent à 20 jusqu'à 40 pour cent des adultes atteints de cancer et sont principalement associés au cancer du poumon et du sein et au mélanome . Ces lésions résultent de la propagation des cellules cancéreuses dans la circulation sanguine et surviennent le plus souvent à la jonction de la matière grise et blanche, où la section transversale des vaisseaux sanguins change, emprisonnant les embolies de cellules tumorales . 80 % des lésions surviennent dans les hémisphères cérébraux, 15 pour cent dans le cervelet et 5 pour cent dans le tronc cérébral. Environ 80 % des patients ont des antécédents de cancer systémique et 70 pour cent ont de multiples métastases cérébrales.

Des progrès significatifs ont récemment été réalisés dans le diagnostic et le traitement de ces lésions, entraînant une amélioration de la survie et du contrôle des symptômes. L'apparition des signes et des symptômes est similaire à celle d'autres lésions massives du cerveau. La méthode de diagnostic de choix est l'imagerie par résonance magnétique utilisant des produits de contraste.
La littérature montre des résultats équivalents pour la chirurgie et la radiochirurgie. Ce dernier semble être plus pratique, efficace et plus sûr pour les petites lésions ou dans les régions inaccessibles à la chirurgie. La radiochirurgie est une alternative judicieuse pour les patients qui ne peuvent pas être opérés pour des raisons médicales. Cependant, la chirurgie est clairement la méthode optimale pour obtenir des tissus pour le diagnostic et pour enlever les lésions qui provoquent un effet de masse. Par conséquent, la radiochirurgie et la chirurgie doivent plutôt être considérées comme deux méthodes complémentaires mais différentes à appliquer en fonction de la situation différente du patient. Pendant près de 50 % des patients présentant une ou deux métastases cérébrales ne sont pas candidats à l'ablation chirurgicale en raison de l'inaccessibilité des lésions, de l'étendue de la maladie systémique ou d'autres facteurs. Ces patients et d'autres atteints de métastases multiples se voient généralement proposer une radiothérapie panencéphalique comme norme de soins. En fait, atteindre jusqu'à près de 50 pour cent d'entre eux avec cette thérapie une amélioration des symptômes neurologiques et 50 jusqu'en 70 pour cent une réaction notable. La chimiothérapie est rarement utilisée principalement pour les métastases cérébrales.
Pour la plupart des patients présentant des métastases cérébrales, la survie médiane n'est que de quatre à six mois après la radiothérapie panencéphalique. Cependant, les patients de moins de 60 ans présentant des lésions discrètes et une maladie systémique contrôlée peuvent avoir une survie plus longue car ils peuvent tolérer une approche thérapeutique plus agressive.
Métastases méningées

Vers 5 pour cent des patients atteints de tumeurs peuvent être diagnostiqués avec des métastases des méninges molles (leptomeninges encephali). Le plus souvent, ils surviennent dans le mélanome, le cancer du sein et du poumon en raison de la propagation des cellules tumorales dans la circulation sanguine. Les cellules malignes se propagent ensuite dans tout le système nerveux central (SNC), généralement via le liquide céphalo-rachidien, communément appelé liquide cérébral.
Un ou plusieurs des signes et symptômes suivants peuvent être causés, entre autres, par des métastases méningées :
- lésions nerveuses locales telles que paralysie des nerfs crâniens, faiblesse motrice et radiculopathies, paresthésies et douleurs,
- invasion directe du cerveau ou du tissu rachidien,
- Trouble des vaisseaux sanguins du cerveau et de la colonne vertébrale avec déficits neurologiques focaux et/ou convulsions,
- Obstacles à l'écoulement normal du liquide céphalo-rachidien avec maux de tête et augmentation de la pression intracrânienne,
- Troubles du fonctionnement normal du cerveau tels que l'encéphalopathie et/ou
- infiltration périvasculaire par des cellules tumorales entraînant des symptômes d'ischémie et d'apoplexie.
Le diagnostic peut être posé par l'examen du liquide céphalo-rachidien ou l'imagerie par résonance magnétique du cerveau et de la moelle épinière. La présence de cellules malignes peut être mesurée à 50 % des patients peuvent être identifiés. Au moins 10 % de patients présentant une atteinte leptoméningée, l'examen cytologique reste négatif. En augmentant le nombre de ponctions lombaires à six et la quantité de volume de liquide retiré à 10 Un millilitre par crevaison peut augmenter la possibilité d'un diagnostic positif. Dans le liquide céphalo-rachidien, la concentration des protéines est généralement élevée, celle du glucose peut être faible en présence de pléiocytose. L'étude radiographique peut montrer une hydrocéphalie sans lésion massive ni hypertrophie diffuse des leptoméninges.
Sans traitement, la survie médiane est de 4 à 6 semaines, le décès étant attribué à une détérioration neurologique progressive. Les métastases leptoméningées sont souvent une manifestation du stade terminal de la maladie principale, et le traitement symptomatique peut être la solution la plus appropriée. Les corticostéroïdes et les analgésiques procurent un soulagement temporaire. Le traitement peut être proposé aux patients présentant une maladie systémique minimale et une condition physique générale acceptable pour soulager les symptômes et prolonger la survie.
La survie médiane peut être améliorée par une radiothérapie des sites symptomatiques et des zones malades plus volumineuses identifiées par radiographie, et par un traitement intrathécal avec méthotrexate, cytarabine et thiotépa, effectué avec une ponction lombaire ou un cathéter Ommaya, à partir de 3 le 6 mois sont augmentés.
La complication majeure du traitement intrathécal à base de méthotrexate est la nécrosantee leucoencéphalopathie, qui peut se développer après des mois de traitement chez les quelques patients qui bénéficient d'une survie prolongée. Cet effet toxique dévastateur est particulièrement fréquent chez les patients qui ont reçu une radiothérapie antérieure ou concomitante avec un traitement au méthotrexate intrathécal.
Douleur et soins terminaux
Les soins palliatifs sont un type particulier de soins fournis pour améliorer la qualité de vie des patients qui souffrent d'une maladie grave ou potentiellement mortelle, comme le cancer. Le but des soins palliatifs n'est pas de guérir mais de prévenir ou de traiter, le plus tôt possible, les symptômes et les effets secondaires de la maladie et de son traitement, ainsi que les problèmes psychologiques, sociaux et spirituels qui y sont liés. Les soins palliatifs sont également appelés soins de confort, soins de soutien et gestion des symptômes.
Les soins palliatifs sont prodigués tout au long de l’expérience d’un patient atteint de cancer. Elle commence généralement au moment du diagnostic et se poursuit tout au long du traitement, des soins de suivi et de la fin de vie.
Liens externes
- www.anocef.org – Association des Neuro-oncologues d'Expression Française (ANOCEF)
Littérature
- (en) Jan C. Buckner et al., Central Nervous System Tumors, , S. 1271–1286
- (en) Lisa M. DeAngelis et al., Intracranial Tumors. Diagnosis and Treatment, London, Dunitz, (ISBN 1-901865-37-1)
- (en) D. N. Louis et al., WHO Classification of Tumours of the Central Nervous System, Genève, , 4e éd. (ISBN 978-92-832-2430-3)
- (en) Richard Pazdur et al., Cancer management. A multidisciplinary approach. Medical, surgical, & radiation oncology, Norwalk, UBM Medica, (ISBN 978-0-615-41824-7)
- (en) Jerome B. Posner, Neurologic Complications of Cancer, Philadelphia, Davis, (ISBN 0-8036-0006-2)
- (de) Rüdiger Schenk, Neuroonkologische Therapiekonzepte zur Behandlung von Astrozytomen höheren Malignitätsgrades und Rezidivlokalisation, Regensburg,
- (de) Uwe Schlegel et al., Neuroonkologie, Stuttgart, Thieme, , 2e éd. (ISBN 3-13-109062-6)
- (en) Jörg-Christian Tonn et al., Oncology of CNS Tumors, Berlin, Springer, , 2e éd. (ISBN 978-3-642-02873-1)