

| Spécialité | Génétique médicale |
|---|
| CIM-10 | Q25.9 |
|---|---|
| CIM-9 | 747.9 |
| OMIM | 608808 |
| DiseasesDB | 13259 |
| MedlinePlus | 001568 |
| eMedicine | 900574 |
| MeSH | D014188 |
| Patient UK | Transposition-of-the-Great-Arteries |
La transposition des gros vaisseaux (TGV) ou discordance ventriculo-artérielle dans la nomenclature actuelle est la malformation cardiaque congénitale cyanogène (responsable d’une cyanose) la plus fréquente chez le nouveau-né. Elle est caractérisée par une malposition des vaisseaux de la base du cœur telle que, à l’inverse du cœur normal, l'aorte est issue du ventricule droit et l'artère pulmonaire du ventricule gauche.
La première description de cette malformation est attribuée à Matthew Baillie (1761-1823) à partir de l'examen anatomique du cœur d'un nourrisson âgé de deux mois (en 1793).
Spontanément, cette cardiopathie est rapidement mortelle dans les quelques jours suivant la naissance mais les différents traitements qui lui sont opposés depuis 1965 en ont radicalement modifié le pronostic, permettant aujourd'hui d'espérer une « guérison complète » dans la grande majorité des cas.
C'est une des malformations cardiaques qui a le plus bénéficié du diagnostic prénatal par échographie fœtale, celui-ci permettant d'organiser au mieux la prise en charge dès la naissance de cette véritable urgence néo-natale.
Fréquence
La TGV représente 5 à 7 % des cardiopathies congénitales. C'est la plus fréquente des cardiopathies cyanogènes du nouveau-né, avant la Tétralogie de Fallot. Son incidence est estimée entre 20 et 30 pour 100 000 naissances, soit 150 à 220 nouveaux cas en France chaque année (sur 783 500 naissances répertoriées par l'INSEE en 2007). Elle touche 2 à 3 fois plus souvent les garçons que les filles.
Anatomie

Dans le cœur normal, l'aorte part du ventricule gauche et distribue le sang riche en oxygène vers l'ensemble du corps alors que l'artère pulmonaire (ou tronc pulmonaire) part du ventricule droit et ramène vers les poumons le sang veineux désaturé, c'est-à-dire appauvri en oxygène.
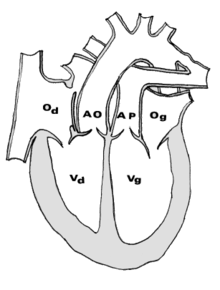
La Transposition des gros vaisseaux est une "malposition vasculaire" dans laquelle l'aorte naît du ventricule droit alors que le tronc pulmonaire naît du ventricule gauche.
Lorsque cette malposition vasculaire est isolée, on parle de transposition simple des gros vaisseaux (l'adjectif simple faisant référence à l'anatomie mais non aux conséquences de la malformation) ou de "D-TGV isolée" en référence à l'anomalie embryologique causale. C'est le cas le plus fréquent (≥ 60 %).
Une D-TGV peut être associée à d'autres malformations cardiaques, en particulier une communication interventriculaire, une sténose sous-pulmonaire ou une coarctation de l'aorte. Certaines de ces anomalies peuvent contribuer à une meilleure tolérance à la naissance, au moins transitoirement, mais toutes tendent à compliquer plus ou moins le traitement chirurgical de la TGV.
Il faut enfin mentionner la possibilité, très rare, d'une double discordance donnant une transposition corrigée des gros vaisseaux. Dans cette malformation, il existe à la fois une discordance ventriculo-artérielle comme dans la TGV simple et une discordance auriculo-ventriculaire telle que l'oreillette droite se draine dans le ventricule gauche et l'oreillette gauche se draine dans le ventricule droit. C'est une malformation anatomiquement plus complexe que celle de la TGV simple mais qui, en pratique, tend à corriger elle-même les anomalies de circulation présentes dans la TGV simple. En effet, en cas de double discordance, le sang veineux désaturé arrivant dans l'oreillette droite rejoint comme normalement le tronc pulmonaire et les poumons (via le ventricule gauche) et le sang riche en oxygène arrivant des poumons dans l'oreillette gauche rejoint comme normalement l'aorte (via le ventricule droit). Les conséquences, complications et traitements éventuels de la transposition corrigée des gros vaisseaux sont radicalement différents de ceux de la TGV simple et ne seront pas présentés dans cet article.
Embryologie

La théorie la plus communément admise aujourd’hui est celle d’un « développement anormal du conus », proposée par Van Praagh.
Le conus est la structure embryologique qui assure la connexion entre les ventricules et les gros vaisseaux qui en partent.
- Normalement, le conus présente une évolution différente dans sa portion sous-pulmonaire, qui grandit et sa partie sous-aortique, qui, elle, a tendance à régresser. Cette résorption du conus sous-aortique entraîne l’anneau aortique vers le bas, l’arrière et la gauche et le place au-dessus du ventricule gauche, en continuité avec l’anneau valvulaire mitral. La persistance du conus sous-pulmonaire déplace l’anneau valvulaire pulmonaire vers le haut, la droite et l’avant, au-dessus du ventricule droit. La torsion du tronc artériel primitif (truncus) secondaire à cette croissance asymétrique est responsable d’un enroulement des vaisseaux de la base et de leur croisement dans l’espace.
- Dans la TGV, c’est le conus sous-pulmonaire qui se résorbe et le conus sous-aortique qui se maintient. L’anneau aortique est donc situé en haut et en avant sur un conus bien développé, au-dessus du ventricule droit, tandis que l’anneau pulmonaire reste au-dessus du ventricule gauche, en continuité avec l’anneau mitral. Les vaisseaux perdent leur enroulement et restent parallèles dans leur portion initiale.
Cette perte du « croisement physiologique » des gros vaisseaux est le signe d’appel échographique principal de l’anomalie.
Physiopathologie
- Avant la naissance, en raison des particularités de la circulation fœtale, cette malformation n'a pratiquement pas de conséquence. La grossesse peut se dérouler normalement avec un accouchement à terme. La croissance fœtale est habituellement normale et il est ainsi classique de dire que les bébés atteints de TGV simple sont de « beaux garçons de plus de 3 kg nés à terme », d’autant plus que l’association à des anomalies extra-cardiaques est rare (moins de 10 % des cas).
- À la naissance, se met en place un mode de circulation sanguine différent, avec deux circuits (cœur droit et cœur gauche) fonctionnant non plus en parallèle mais en série. Les deux circuits ne sont alors plus interchangeables mais strictement séparés et le sang doit les emprunter successivement, l'un puis l'autre. En cas de TGV, le sang désaturé en oxygène arrivant à l'oreillette droite repart directement d'où il vient via l'aorte vers tout le corps. Le sang riche en oxygène arrivant à l'oreillette gauche en fait de même en retournant directement vers les poumons par le tronc pulmonaire. Les deux circuits sont séparés et indépendants : le sang désaturé ne rejoint pas les poumons et le sang riche en oxygène ne parvient plus au corps.
La survie immédiate est conditionnée par la persistance des communications (shunts) qui existaient entre les deux circuits avant la naissance : le foramen ovale (entre les oreillettes) et le canal artériel (entre aorte et artère pulmonaire). Elles seules permettront un mélange des sangs désaturé et oxygéné. Or, ces shunts fœtaux sont destinés à se fermer à la naissance, dans un délai non prévisible allant de quelques minutes à 48-72 heures. La TGV est donc une cardiopathie « ducto-dépendante » et surtout « atrio-dépendante » (l'apport en sang oxygéné dans le cœur droit et l'aorte est lié essentiellement à la présence d'une communication entre les oreillettes) et son traitement est une réelle urgence néo-natale.
Les formes associées à une communication inter-ventriculaire (CIV) sont souvent mieux tolérées à la naissance dans la mesure où cette CIV, surtout si elle est large, ne se fermera pas aussi rapidement et permettra un meilleur mélange sanguin, mais au prix d'une insuffisance cardiaque. Historiquement, les seules formes qui pouvaient survivre spontanément quelques mois ou quelques années étaient les TGV associées à une large CIV et à une sténose (rétrécissement) sous-pulmonaire modérée, cette dernière limitant l'importance de l'insuffisance cardiaque.
Diagnostic
Anténatal
Le diagnostic de TGV est de plus en plus fréquemment porté avant la naissance, le plus souvent lors de l'examen échographique « morphologique » fait à 22 semaines d'aménorrhée (SA). Il est parfois possible plus tôt, dès 12-14 SA, quand les conditions d'examen sont particulièrement favorables (... et l'examinateur expérimenté).
Aspects échographiques avant la naissance



Les structures strictement intra cardiaques étant habituellement normales, le diagnostic ne peut être évoqué sur l'incidence dite des "4 cavités". Il nécessite obligatoirement (et logiquement) une exploration des gros vaisseaux passant par d'autres plans de coupe plus haut situés dans le thorax.
- le signe d'appel habituel est la perte du croisement des gros vaisseaux. Normalement, l'aorte et le tronc pulmonaire se croisent peu après leur sortie du cœur. Il est donc impossible de dérouler les deux vaisseaux sur un même plan de coupe. Quand l'aorte est vue longitudinalement, le tronc pulmonaire n'apparaît qu'en section transversale ("aorte en long, tronc pulmonaire en rond") et vice-versa. Dans la D-TGV simple, la plus fréquente, les vaisseaux ont un trajet parallèle et peuvent donc être visualisés longitudinalement simultanément.
- la confirmation est obtenue par l'analyse des gros vaisseaux et de leurs connexions aux ventricules. L'aorte est caractérisée par le fait qu'elle décrit une crosse, donne naissance aux vaisseaux à destinée céphalique et est le vaisseau qui monte le plus haut dans le thorax. Le tronc pulmonaire est caractérisé par sa bifurcation précoce en deux branches, les artères pulmonaires droite et gauche. Sur un cœur par ailleurs normal, le ventricule droit est situé en avant et à droite du ventricule gauche, le tronc pulmonaire est donc le vaisseau le plus antérieur. La TGV se caractérise par la position antérieure du vaisseau qui décrit la crosse et donne les vaisseaux céphaliques : l'aorte.
Gestion de la grossesse
Le diagnostic de TGV simple ne doit pas modifier le cours de la grossesse mais peut indiquer une surveillance échographique plus étroite et surtout incite à préparer un accouchement (programmé) à proximité d'un centre de cardiologie pédiatrique apte à assurer la prise en charge néo-natale (voir plus bas).
- Poursuite de la grossesse : En l'absence d'anomalie, cardiaque ou extra-cardiaque associée, le risque d'anomalie génétique identifiable est considéré comme très faible sinon nul. Il n'y a donc pas d'indication formelle à la réalisation d'un prélèvement de liquide amniotique (amniocentèse). La croissance et le développement du fœtus ne sont habituellement pas perturbés. Après explications et compte tenu du pronostic postopératoire en règle excellent, il est exceptionnel que les parents formulent une demande d'interruption médicale de grossesse, laquelle serait d'ailleurs fortement discutée par le centre de diagnostic prénatal impliqué.
- Surveillance échographique : Celle-ci poursuit plusieurs buts :
- soutien psychologique et réponse aux interrogations des parents,
- confirmation d'une croissance normale et de l'absence d'anomalie associée (certaines peuvent n'être dépistées que tardivement, au 3e trimestre)
- étude détaillée de certaines données anatomiques (en particulier diamètres des gros vaisseaux) ou fonctionnelles (présence d'un foramen ovale restrictif) qui pourraient influer sur la tolérance néo-natale ou la qualité du résultat chirurgical.
- Accouchement : Celui-ci peut se dérouler normalement, par voie basse, le recours à une césarienne n'étant pas obligatoire. L'accouchement est cependant programmé dans un centre réunissant les compétences obstétricales, de cardiologie pédiatrique interventionnelle, de chirurgie cardiaque et réanimation néo-natale permettant une prise en charge optimale du nouveau-né.
Postnatal
Clinique
La transposition des gros vaisseaux doit être évoquée en premier lieu chez un nouveau né qui présente une cyanose (coloration bleutée de la peau), et ceci d'autant plus que :
- la cyanose est isolée, sans détresse respiratoire, sans souffle cardiaque réel, sans insuffisance cardiaque (au moins dans les premières heures de vie) ;
- cette cyanose n'est pas (ou que très peu) modifiée par l'administration d'oxygène au nouveau né (cyanose réfractaire à l'oxygénothérapie).
Après quelques heures d'évolution peuvent apparaître des signes de défaillance cardiaque (polypnée, tachycardie, marbrures, sueurs, refus du biberon...).
Examens complémentaires
Les examens permettant de poser le diagnostic de TGV sont essentiellement :
- La radiographie du thorax de face qui peut montrer une silhouette cardiaque évocatrice et surtout une vascularisation pulmonaire normale ou augmentée tout à fait inattendue dans une cardiopathie cyanogène.
- L'échocardiographie qui est de plus en plus l'examen pratiqué en première intention. Cet examen permet d'affirmer le diagnostic en montrant les anomalies anatomiques, d'apprécier le caractère plus ou moins fonctionnel des shunts cardiaques fœtaux, de rechercher d'autres anomalies associées et dans une certaine mesure d'apprécier le retentissement de la malformation.
- Les coupes échographiques les plus utiles à cette fin sont :



- la coupe parasternale gauche grand axe : elle montre que le vaisseau issu du ventricule gauche (le plus postérieur) se dirige anormalement vers l'arrière et se bifurque précocement. C'est donc le tronc pulmonaire et non l'aorte comme normalement ;
- les coupes sous costales qui montrent bien le trajet parallèle des gros vaisseaux et leur inversion.
- Cet examen permettra d'éliminer en particulier un retour veineux pulmonaire anormal total bloqué qui aurait pu rendre compte de l’hypervascularisation pulmonaire sur la radiographie des poumons.
- Un cathétérisme cardiaque n'est plus, sauf exception, réalisé à titre diagnostique mais reste nécessaire pour la réalisation d'un geste thérapeutique, l'atrioseptostomie de Rashkind.
Traitement
Sans chirurgie, la mortalité dépasse 90 % à un an. Elle est inférieure à 10% à 20 ans en cas de réparation.
Prise en charge du nouveau-né
Idéalement, c'est-à-dire quand le diagnostic a été fait avant la naissance, l'accouchement ne peut se concevoir que dans une maternité disposant d'un service de cardiologie pédiatrique. La prise en charge débute immédiatement après la naissance, en salle de travail ou à proximité immédiate. Elle associe le plus souvent :
- une confirmation du diagnostic par examen échocardiographique. Celui-ci permet également de faire le bilan des anomalies éventuellement associées et des particularités anatomiques et fonctionnelles du foramen ovale et du canal artériel ;
- la mise en place d'une perfusion de prostaglandine (PGE1), médicament qui inhibe la vasoconstriction du canal artériel et permet de le maintenir ouvert (éventuellement plusieurs jours) ;
- la réalisation d'une « atrioseptostomie » par la manœuvre de Rashkind (voir plus bas). Celle-ci, pas toujours nécessaire, vise à maintenir le shunt qui existait par le foramen ovale en créant une (large) communication inter-auriculaire "permanente".
- Lorsque le diagnostic de TGV n'est soupçonné qu'après la naissance, les deux premiers éléments de la prise en charge sont habituellement assurés par le service de néonatalogie le plus proche de la maternité de naissance, puis le nouveau-né est secondairement transféré en milieu cardiopédiatrique spécialisé pour la poursuite du traitement.
- La « manœuvre de Rashkind » ou atrioseptostomie de Rashkind a été décrite initialement en 1966 par Rahskind. C'est le premier traitement (palliatif) non chirurgical qui ait été proposé pour la TGV. C'est également la première des interventions dites par « cathétérisme interventionnel ». Elle consiste à introduire par la veine fémorale ou la veine ombilicale, une sonde munie d'un ballonnet gonflable à son extrémité. Cette sonde est guidée dans l'oreillette droite puis, à travers le foramen ovale, poussée dans l'oreillette gauche. Le ballonnet est alors gonflé puis, d'un geste sec, la sonde est retirée de quelques centimètres. Le ballon passe ainsi de l'oreillette gauche à l'oreillette droite en déchirant la cloison inter-auriculaire, créant ainsi une communication permanente. Plusieurs passages du ballon peuvent être nécessaires pour créer une communication suffisamment large, l'efficacité de la manœuvre étant jugée instantanément par la mesure de la saturation en oxygène dans le sang artériel (ou capillaire). Cette manœuvre, qui nécessite un cathétérisme se pratiquait à l'origine dans une salle spéciale, munie d'un appareillage radiologique adapté. Actuellement, elle se fait sous simple surveillance échographique et peut être réalisée en salle de travail ou dans l'incubateur du nouveau-né.
Traitement chirurgical
Le traitement définitif de la TGV est chirurgical. Il a considérablement évolué au fil du temps, tant pour le mode de « correction » que pour l'âge auquel celle-ci est pratiquée. Actuellement, il est courant sinon habituel qu'un nouveau-né soit de retour à domicile à 15 jours de vie, « guéri » et sans aucun traitement médical après « détransposition des gros vaisseaux. »
Historiquement, trois types de chirurgie ont été proposés, les deux premiers pouvant encore être d'actualité dans des cas très particuliers.
- L'intervention de Blalock-Hanlon (1950) : C'est la première intervention chirurgicale (palliative) proposée dans la transposition des gros vaisseaux. Elle consistait en la création, sur cœur battant et sans circulation extra-corporelle d'une large communication inter auriculaire visant à améliorer l’oxygénation du sang circulant dans le ventricule droit et l’aorte.
Après 1966, elle est restée indiquée en cas d'échec ou d'insuffisance de la manœuvre de Rashkind ou pratiquée de façon plus ou moins systématique dans certaines écoles comme celle de Paris pour permettre d’atteindre un âge plus avancé (12-24 mois) avant l’intervention définitive de "correction à l'étage auriculaire".
- Les corrections physiologiques à l'étage auriculaire : Le principe de ces interventions, faussement qualifiées de correctrices, était non pas de remettre les gros vaisseaux en place normale mais de transformer la « transposition simple des gros vaisseaux » en « transposition corrigée des gros vaisseaux », ceci en inversant les flux sanguins au niveau des oreillettes.

Par un néo-cloisonnement des oreillettes, le sang veineux en provenance des veines caves était redirigé vers l'oreillette gauche, le ventricule gauche et l'artère pulmonaire; le sang oxygéné venant des poumons étant détourné vers l'oreillette droite, le ventricule droit et l'aorte. Une circulation sanguine "normale" était ainsi restituée, mais non une anatomie normale puisque les gros vaisseaux restaient connectés au « mauvais » ventricule. Deux chirurgiens ont associé leurs noms à ce type de correction : Senning en 1959 et Mustard en 1964 et ces interventions ont été pratiquées jusqu'aux années 1975-80. Malgré certaines complications liées aux lésions créées au niveau des oreillettes (risque de troubles du rythme ou de la conduction, de sténose des chenaux cave ou pulmonaire, surtout avec le procédé décrit par Mustard qui reposait sur l’emploi d’un patch inerte et donc incapable de suivre la croissance de l’enfant) et au maintien du ventricule droit en position « systémique » c’est-à-dire sous l’aorte (risque de défaillance à long terme), de nombreux enfants ainsi opérés sont maintenant des adultes menant une vie sensiblement normale. Dans ses dernières années, William Mustard se faisait souvent accompagner dans les congrès médicaux par une jeune femme, mère de famille, qui était sa première ou une de ses premières opérées. En France, de nombreux adultes doivent leur salut au chirurgien George Lemoine, « homme modeste, trop peu connu » (Y. Lecompte) qui n’avait pas son pareil pour réaliser une intervention de Senning vers l’âge de 6 mois.
- La correction anatomique à l'étage artériel : Corriger la TGV en remettant les vaisseaux en place normale est la logique même et dès 1954, plusieurs chirurgiens s’y sont essayés (Mustard, Bjork, Bailey, Kay et Cross, Baffes …) mais sans succès. Ces échecs tenaient aux limites techniques de l’époque. C’était les débuts de la chirurgie à cœur ouvert chez l’adulte et la préhistoire de cette chirurgie chez le nouveau-né ou le nourrisson. Ils tenaient également à la sous-estimation de certaines contraintes physiologiques : les artères coronaires irriguant le muscle cardiaque étaient laissées en place sur l’artère pulmonaire et donc perfusées à trop basse pression par un sang désaturé. Par ailleurs, le ventricule gauche, déshabitué à pousser contre des résistances élevées, présentait une défaillance irréversible dès qu’il était branché sur l’aorte. Ces échecs initiaux expliquent l’intérêt porté aux interventions de correction à l’étage auriculaire dans les 20 années qui ont suivi. Elles étaient certes moins logiques mais efficaces.
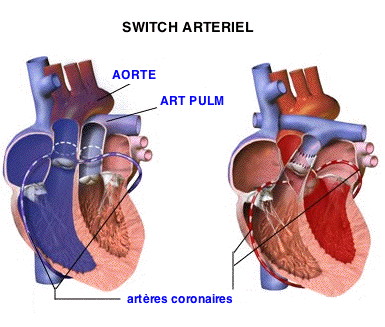
Les tentatives reprirent 20 ans plus tard, au milieu des années 1970 après la publication par le chirurgien brésilien Adib Jatene d'une chirurgie de détransposition pratiquée avec succès chez deux enfants âgés de 3 mois et 40 jours et les premières séries opératoires de Yacoub en Angleterre ainsi que de l’équipe de l’hôpital Laennec à Paris. Ces nouvelles tentatives visaient à corriger les défauts des premières, d’une part en transposant également les artères coronaires sur l’aorte, d’autre part en veillant à l’aptitude du ventricule gauche à assurer un débit systémique. Cette aptitude était obtenue soit par une « préparation » du ventricule, par mise en place quelques mois d’un cerclage de l’artère pulmonaire l’obligeant ainsi à se muscler pour vaincre cet obstacle artificiel, soit par un timing opératoire très précoce, dès la première semaine de vie, avant que le myocarde ventriculaire gauche n’ait significativement involué. La détransposition des gros vaisseaux ou « switch artériel » devint rapidement l’intervention de référence après que Yacoub a codifié les techniques permettant d’adapter l’intervention aux différentes distributions possibles des artères coronaires et que Lecompte a « inventé » en 1979 un procédé de décroisement des gros vaisseaux permettant une réimplantation de l’artère pulmonaire sans interposition de matériel prothétique, reconnu depuis de façon universelle comme « la manœuvre de Lecompte »…
C’est maintenant la seule intervention pratiquée, sauf cas très particulier. C'est réellement une chirurgie correctrice puisqu'elle "détranspose" les gros vaisseaux en réimplantant l'aorte sur le ventricule gauche et l'artère pulmonaire sur le ventricule droit. La mortalité hospitalière est actuellement inférieure à 1 % pour les équipes pratiquant cette intervention couramment et pourtant, il s'agit d'une véritable prouesse de chirurgie et de réanimation néo-natale et cette intervention sous circulation extra-corporelle a même pu être pratiquée sur des prématurés de 2000 grammes à peine.
Stratégies thérapeutiques
Dans les pays développés, la stratégie « idéale » actuelle, de plus en plus souvent « réelle » peut être résumée comme suit : Diagnostic ante-natal et transfert in utero dans un centre spécialisé permettant la pratique de la manœuvre de Rashkind et la perfusion de prostaglandines (non systématiques). La détransposition est faite alors dans la première semaine de vie permettant un retour à domicile dans le premier mois.
Dans les pays en voie de développement, cette stratégie « idéale » nécessite un environnement chirurgical et de réanimation néo-natal hors de portée. Le diagnostic ante-natal de TGV (rarement posé) conduit encore souvent à une interruption médicale de grossesse. On pourrait pourtant y concevoir une réhabilitation des séquences de prise en charge « historique », avec deux options : une manœuvre de Rashkind (associée éventuellement à une intervention de Blalock-Hanlon) suivie d'une correction à l’étage auriculaire par l’intervention de Senning entre 6 et 18 mois, ou une manœuvre de Rashkind (avec éventuelle intervention de Blalock-Hanlon) associée à un cerclage de l’artère pulmonaire, suivie d'une correction anatomique par détransposition entre 6 mois et un an. Ces stratégies étaient celles pratiquées en France il n'y a guère plus de 20 ans et nombreux sont les nourrissons, aujourd'hui adultes, à en avoir réellement bénéficié.
Devenir des enfants
Les enfants opérés par détransposition des gros vaisseaux mènent habituellement une vie strictement normale en l'absence de tout traitement médical. Seuls les sports de compétition ainsi que le tabac restent déconseillés. Ces enfants doivent cependant faire l'objet d'un suivi médical régulier (annuel le plus souvent) pour surveiller plus particulièrement :
- l'absence de complication liée à la réimplantation des artères coronaires. Cette surveillance peut passer par la réalisation d'une coronarographie ou, plus souvent maintenant d'une exploration par tomodensitométrie (scanner coronaire) ;
- l'absence de complication au niveau des sutures artérielles (sténoses, rares et habituellement précoces) ou des valves aortiques et pulmonaires (une petite fuite sans conséquence est habituelle).
Grossesse
Sauf cas particulier (et rare), une femme opérée dans l'enfance d'une transposition des gros vaisseaux par switch artériel peut envisager de mener à bien une ou plusieurs grossesses sans souci particulier. L'accouchement pourra se faire normalement, par voie basse, sauf indication obstétricale à une césarienne. Un allaitement sera possible.
L'inquiétude parentale devant le risque de récidive de la cardiopathie, de l'ordre de 10 % (ce qui signifie 'a contrario' que 90 % au moins des grossesses donneront naissance à un enfant normal) pourra être levée par la réalisation précoce d'une échographie fœtale, dès la 14-15e semaine d'aménorrhée quand les conditions d'examen sont bonnes.
Conseil génétique
Jusqu'à présent, il n'a pas été mis en évidence de cause génétique (ou toxique) à cette malformation quand elle est isolée. À ce titre, c'est l'une des très rares malformations cardiaques qui ne soient pas une indication reconnue d'amniocentèse (ponction du liquide amniotique en vue d'une étude du caryotype fœtal) lorsqu'elle est découverte avant la naissance.
Il en va tout autrement si la transposition des gros vaisseaux est associée à d'autres malformations, cardiaques ou extra-cardiaques. Dans ce cas, la crainte qu'une anomalie génétique soit responsable ou au moins associée à ce syndrome polymalformatif conduit habituellement à proposer aux parents la réalisation d'une amniocentèse avant la naissance ou d'une étude directe du caryotype du nouveau-né si le diagnostic est plus tardif..
Sources
- Heart disease in Infants, Children and Adolescents - Mosse and Adams - 7e édition - Williams & Wilkins éd. - Philadelphie, 2008
- Fetal cardiology - S. Yagel, N.H. Silverman, U. Gembruch - Martin Dunitz ed. - Londres, 2003