
| Syndrome de Protée | |
| Référence MIM | 176920 |
|---|---|
| Transmission | Dominante |
| Chromosome | 14q32.32 |
| Gène | PIK3CA |
| Empreinte parentale | Non |
| Porteur sain | sans objet |
| Nombre de cas | 120 cas environ en 1 988. |
| Maladie génétiquement liée | Syndrome de Cowden-Syndrome de Bannayan-Riley-Ruvalcaba |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
|
modifier |
|
Le syndrome de Protée est une maladie génétique complexe comprenant des hamartomes de taille importante impliquant plusieurs tissus : tissu conjonctif, tissu épidermique et tissu osseux.
Elle se manifeste dès la naissance et les hamartomes grandissent au cours de la vie. Les tumeurs malignes sont rares. Des tumeurs de l'ovaire, des tumeurs testiculaires, des tumeurs de la parotide et des tumeurs du système nerveux sont parfois associées avec les hamartomes.
Son nom provient de la divinité grecque Protée.
Historique
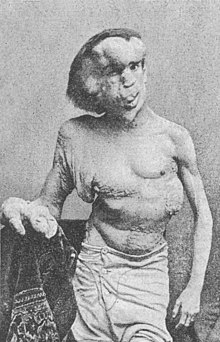
Le syndrome est individualisé pour la première fois en 1976 mais n'est nommé ainsi qu'en 1983.
Joseph Merrick, connu sous le surnom de l'homme éléphant, était très probablement porteur de cette pathologie et non pas de la maladie de Recklinghausen, ni de l'éléphantiasis. Des tests d'ADN récents le prouveraient, comme les recherches le soupçonnaient depuis longtemps.
Cause
Le syndrome de Protée résulte en une insuffisance d'apoptose. Il s'agit d'une mutation activatrice de type mosaïque où le génome des lésions (hamartome) est différent de celui du tissu normal chez le même sujet. Une mutation sur l'oncogène AKT1 (codant la protéine kinase B ou PKB ou AKt) est retrouvée uniquement dans le tissu des lésions. Cette mutation entraîne une phosphorylation perpétuelle de la PKB par le phosphatidylinositol-3,4,5-trisphosphate ou PIP3, la rendant active, favorisant ainsi la prolifération cellulaire. En effet, la PKB est capable d'inhiber toute une série de molécules (la cycline D1, la protéine BAD, mTOR via TSC1 et TSC2, FOXP2), impliquées dans la survie, la progression du cycle cellulaire,
Liens externes
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:176920 [1]
- Ressources relatives à la santé :
- Orphanet
- (en) Diseases Ontology
- (en) DiseasesDB
- (en + es) Genetic and Rare Diseases Information Center
- (en) Héritage mendélien chez l'humain
- (en) Héritage mendélien chez l'humain
- (en) Medical Subject Headings
- (en) NCI Thesaurus
- Notice dans un dictionnaire ou une encyclopédie généraliste :