
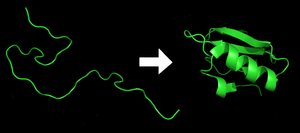
Le repliement des protéines est le processus physique par lequel un polypeptide se replie dans sa structure tridimensionnelle caractéristique dans laquelle il est fonctionnel.
Chaque protéine commence sous forme de polypeptide, transcodée depuis une séquence d'ARNm en une chaîne linéaire d'acides aminés. Ce polypeptide ne possède pas à ce moment de structure tridimensionnelle développée (voir côté gauche de la figure). Cependant, chaque acide aminé de la chaîne peut être considéré comme ayant certaines caractéristiques chimiques essentielles. Cela peut être l'hydrophobie, l'hydrophilie, ou la charge électrique, par exemple. Elles interagissent entre elles et ces interactions conduisent, dans la cellule, à une structure tridimensionnelle bien définie, la protéine repliée (à droite sur la figure), connue comme l'état natif. La structure tridimensionnelle résultante est déterminée par la séquence des acides aminés. Le mécanisme du repliement des protéines n'est pas encore complètement compris, en particulier l'ordre dans lequel les différentes parties se replient. Le problème est ardu car, par exemple, certaines parties déjà repliées aident au repliement d'autres parties, ce qui rend le problème non linéaire.
La détermination expérimentale de la structure tridimensionnelle d'une protéine est souvent très difficile et coûteuse. Cependant, la séquence de cette protéine est connue, en particulier depuis le séquençage complet de génomes et la détection automatiques de séquences codantes. En conséquence, les scientifiques ont essayé d'utiliser plusieurs techniques biophysiques pour replier « manuellement » une protéine, c'est-à-dire de prédire la structure d'une protéine complète à partir de sa séquence. Si cette méthode a apporté des résultats intéressants avec de courtes protéines, l'état actuel de la science achoppe complètement à prédire la structure tridimensionnelle des protéines intégrales de membranes. D'autres protéines échappent à cette analyse, par exemple les protéines possédant de nombreux ponts disulfures ou encore des protéines synthétisées sous forme de pré-protéine, c'est-à-dire sous forme de protéine précurseur clivée par des protéases spécifiques pour acquérir leur maturité. C'est le cas par exemple de l'insuline.
La structure tridimensionnelle correcte, ou native, est essentielle pour que la protéine puisse assurer sa fonction au sein de la cellule. L'échec du repliement dans la forme attendue produit des protéines inactives avec des propriétés différentes (par exemple, le prion). De nombreuses maladies neurodégénératives ou autres sont considérées comme résultant d'une accumulation de protéines « mal repliées ».
Historique
Christian Boehmer Anfinsen démontre en 1961 le repliement de la ribonucléase et postule que la conformation finale dépend essentiellement de la succession d'acides aminés qui constitue la protéine. Il reçoit en 1972 le prix Nobel de chimie. Les protéines chaperons, nécessaires à certains repliements, ont été découvertes à la fin des années 1980 par Franz-Ulrich Hartl et Arthur Horwich. Les deux scientifiques reçoivent en 2011 le Prix Albert-Lasker pour la recherche médicale fondamentale pour leurs travaux sur le sujet.
Toutefois, ce dogme repose sur l'idée que le repliement ne dépend que de contraintes thermodynamiques. Par la suite, à partir du modèle d'allostérie développé par Monod-Wyman et Changeux, Jeannine Yon et de Michel Goldberg, dans des travaux menés en parallèle, introduisent progressivement en France l'idée d'une contrainte cinétique jouant aussi un rôle dans ces repliements.
Connaissances actuelles du processus
Relation entre repliement et séquence d'acides aminés
La séquence d'acides aminés (ou structure primaire) d'une protéine la prédispose à adopter sa conformation native (ou une de ses conformations natives). Elle se repliera spontanément pendant ou après sa synthèse. Alors que ces macromolécules peuvent être considérées comme se « repliant elles-mêmes », le mécanisme dépend également des caractéristiques du cytosol, comme la nature du solvant primaire (eau ou lipide), la concentration de sels, la température, et des protéines chaperonnes.
La plupart des protéines repliées possèdent un cœur hydrophobe dans lequel l'ensemble des chaines latérales hydrophobes stabilisent l'état replié, et des chaînes latérales polaires ou chargées sur leur surface exposée au solvant par lesquelles elles interagissent avec les molécules d'eau environnantes. Il est généralement admis que la minimisation du nombre de chaînes latérales hydrophobes exposées à l'eau est la principale force motrice du processus de repliement, bien qu'une théorie récemment proposée mette l'accent sur les contributions apportées par la liaison hydrogène.
Le processus de repliement in vivo débute parfois lors de la traduction, c'est-à-dire que la terminaison N de la protéine commence à se replier alors que la portion terminale C de la protéine est toujours en cours de synthèse par le ribosome. Les protéines spécialisées appelées chaperonnes aident au repliement des autres protéines. Le système bactérien GroEL, qui aide au repliement des protéines globulaires, est un exemple bien étudié. Dans les organismes eucaryotes, les protéines chaperonnes sont connues sous le nom de protéines de choc thermique. Bien que la plupart des protéines globulaires soient capables d'atteindre leur état natif sans assistance, les repliements assistés par les protéines chaperonnes sont parfois nécessaires dans un environnement intracellulaire encombré afin de prévenir l'agrégation ; les protéines chaperonnes sont aussi utilisées pour empêcher les mauvais repliements et les agrégations pouvant se produire en conséquence d'une exposition à la chaleur ou à d'autres changements dans l'environnement cellulaire.
De nombreux scientifiques ont été capables d'étudier plusieurs molécules identiques se repliant ensemble de manière massive. Au niveau le plus basique, il apparaît que lors de la transition vers l'état natif, une séquence d'acides aminées donnée prend à peu près le même chemin et utilise à peu près les mêmes intermédiaires et états de transition. Le repliement implique parfois la création de structures secondaires et supersecondaires régulières, particulièrement les hélices alpha et les feuillets bêta, puis de la structure tertiaire. La formation de la structure quaternaire implique l'« assemblage » ou le « coassemblage » de sous-unités qui se sont déjà repliées. Les structures d'hélice alpha et de feuillet bêta régulières se replient rapidement car elles sont stabilisées par des liaisons hydrogène, comme l'a établi en premier Linus Pauling. Le repliement protéique peut impliquer des liaisons covalentes sous la forme de ponts disulfures formés entre deux résidus de cystéine ou la formation de clusters métalliques. Peu avant d'occuper leur conformation native énergétiquement favorable, les molécules peuvent passer par un état intermédiaire de globule fondu.
Le point essentiel du repliement, cependant, reste que la séquence d'acides aminés de chaque protéine contient l'information spécifiant à la fois la structure native et le chemin pour y accéder. Ce qui ne veut pas dire que deux séquences d'acides aminés identiques se replient à l'identique. Les conformations diffèrent selon les facteurs environnementaux par exemple; des protéines similaires se replient différemment selon l'endroit où elles se trouvent. Le repliement est un processus spontané indépendant de l'apport énergétique des nucléosides triphosphates. Le passage à l'état replié est principalement guidé par les interactions hydrophobes, la formation de liaisons hydrogène intramoléculaires et les forces de Van der Waals, et est contrarié par l'entropie conformationnelle, qui peut être surmontée par des facteurs extrinsèques comme les protéines chaperonnes.
Écart à l'état natif

Dans certaines solutions et sous certaines conditions les protéines ne peuvent se replier dans leurs formes biochimiques fonctionnelles. Des températures au-dessus (et parfois en dessous) de l'intervalle dans lequel les cellules vivent causeront le non-repliement des protéines, ou leur dénaturation (c'est une des raisons pour lesquelles le blanc d'œuf est opaque après avoir bouilli). Des fortes concentrations de solutés, des valeurs de pH extrêmes, des forces mécaniques appliquées, ou encore la présence de dénaturants chimiques peuvent conduire au même résultat. Une protéine complètement dénaturée ne possède ni structure tertiaire ni structure secondaire, et existe sous forme de pelote aléatoire. Sous certaines conditions, certaines protéines peuvent se replier à nouveau ; cependant, dans de nombreux cas la dénaturation est irréversible. Les cellules protègent parfois leurs protéines contre l'influence de la chaleur avec des enzymes connues sous le nom de chaperonnes ou protéines de choc thermique, qui aident les autres protéines à la fois à se replier et à rester pliées. Certaines protéines ne se replient jamais dans les cellules sans l'aide des protéines chaperonnes, qui sont en mesure d'isoler les protéines les unes des autres, ce qui fait que leur repliement n'est pas interrompu par les interactions avec les autres protéines. Elles peuvent aussi aider à déplier les protéines mal repliées, en leur donnant une autre chance de se replier correctement. Cette fonction est cruciale pour prévenir du risque de précipitation en agrégats amorphes insolubles.
Repliements de protéines erronés et maladies neurodégénératives
Les protéines mal repliées sont responsables des maladies liées au prion comme la maladie de Creutzfeldt-Jakob, l'encéphalopathie spongiforme bovine (ou maladie de la vache folle), les maladies de type amylose comme la maladie d'Alzheimer, et de nombreuses autres formes de protéopathie comme la fibrose cystique. Ces maladies sont associées à la multimérisation des protéines non repliées dans les agrégats extracellulaires ou les inclusions intracellulaires insolubles ; il n'est pas établi si les plaques constituent une cause ou un symptôme de la maladie.
Cinétique et paradoxe de Levinthal
La durée globale du procédé de repliement varie drastiquement selon la protéine que l'on considère. Les repliements les plus lents demandent de plusieurs minutes à plusieurs heures pour se produire, principalement en raison des isomérisations de proline ou de mauvaises formations de liaisons disulfures, et la plupart transitent par des états intermédiaires, un peu comme des points de contrôle, avant que le processus soit achevé. D'un autre côté, les très petites protéines à simple domaine avec des longueurs allant jusqu'à une centaine d'acides aminés se replient en une seule étape. Des échelles de temps de quelques millisecondes constituent la norme et les réactions de repliement des protéines les plus rapides connues se produisent en quelques microsecondes. Le paradoxe de Levinthal indique que si une protéine se replie en échantillonnant toutes les conformations, cela prendrait une durée de temps astronomique pour le faire, même si les conformations étaient échantillonnées à vitesse rapide (de l'échelle de la nanoseconde ou de la picoseconde). En se basant sur l'observation du fait que les protéines se replient bien plus rapidement que ça, Cyrus Levinthal a proposé qu'une recherche conformationnelle aléatoire ne se produit pas durant le repliement, et que la protéine doit, plutôt, se replier selon un « chemin » préférentiel.
Techniques d'études du repliement des protéines
Études récentes du repliement avec une haute résolution en temps
L'étude du repliement des protéines a été très largement amélioré dans ces dernières années par le développement des techniques disposant d'une puissante résolution temporelle. Ce sont des méthodes expérimentales pour déclencher rapidement le repliement d'une protéine, puis observer la dynamique résultante. Les techniques rapides en usage large comprennent le mélange ultra-rapide des solutions, des méthodes photochimiques, et la spectroscopie de saut de température par laser. Parmi les nombreux scientifiques ayant contribué au développement de ces techniques, on trouve Heinrich Roder, Harry Gray, Martin Gruebele, Brian Dyer, William Eaton, Sheena Radford, Chris Dobson, Alan Fersht et Bengt Nölting.
Théorie du paysage d'énergie du repliement des protéines
Le phénomène de repliement des protéines fut principalement un effort expérimental jusqu'à l'énoncé de la théorie du paysage d'énergie par Joseph Bryngelson et Peter Wolynes à la fin des années 1980 et au début des années 1990. Cette approche introduit le principe de moindre frustration qui spécifie que l'évolution a sélectionné les séquences d'acides aminés dans les protéines naturelles de sorte que les interactions entre les chaînes latérales favorisent l'acquisition par la molécule de son état replié. Les interactions qui ne favorisent pas ce repliement sont identifiées comme telles et « désélectionnées », bien que de la « frustration » résiduelle soit attendue. Une des conséquences de la sélection de ces séquences par l'évolution est que ces protéines sont généralement censées avoir un processus de repliement au sein d'un « paysage d'énergie orienté » (selon l'expression de José Onuchic) qui pointe largement vers l'état natif. Cette direction de repliement du paysage d'énergie autorise la protéine à se replier vers l'état natif via n'importe lequel des chemins et des intermédiaires, plutôt que d'être restreint à un seul mécanisme. Cette théorie est appuyée par des simulations numériques de protéines modèles et a été utilisée pour la prédiction de structures et en conception de protéines.
Prédiction numérique de la structure tertiaire des protéines
Les techniques de novo ou ab initio pour la prédiction numérique de structures protéiques sont liées, mais distinctes, aux études sur le repliement des protéines. La dynamique moléculaire (DM) est un outil important pour l'étude du repliement et de la dynamique des protéines in silico. En raison du coût numérique, les simulations de repliements par dynamique moléculaire ab initio avec de l'eau explicite sont limitées à des peptides et des très petites protéines. Les simulations DM de protéines plus grosses restent restreintes aux dynamiques sur la structure expérimentale ou sa structure non-repliée à haute température. Afin de simuler les processus de repliements longs (au-delà d'une microseconde environ), comme le repliement des protéines de petites tailles (environ 50 résidus) ou plus grosses, des approximations ou des simplifications des modèles de protéines doivent être introduites. Une approche utilisant des représentations réduites des protéines (des pseudo-atomes représentant des groupes d'atomes sont définis) et des potentiels statistiques ne sont pas seulement utiles dans l'optique d'une prédiction de structure protéique, mais sont aussi capables de reproduire les chemins de repliements.
En raison des plusieurs voies possibles de repliement, il peut exister plusieurs structures possibles. Un peptide constitué de seulement cinq acides aminés peut se replier en plus de 100 milliards de structures potentielles
.
Techniques de détermination des structures de protéines
La détermination de la structure repliée d'une protéine est une procédure longue et complexe, impliquant des méthodes comme la diffractométrie de rayons X ou la RMN. Un des champs de plus grand intérêt est la prédiction des structures natives à partir des seules séquences d'acides aminés en utilisant la bio-informatique et des méthodes de simulations numériques.
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Protein folding » (voir la liste des auteurs).
Voir aussi
Articles connexes
- Protéines intrinsèquement désordonnées
- Agrégation des protéines
- Paradoxe Levinthal
- Dénaturation
- Modélisation moléculaire
- Folding@Home
- Blue Gene
- Foldit, jeu vidéo sur le repliement des protéines
Liens externes
- (en) Folding @ Home Aide au décodage du repliement des protéines.
- (en) Rosetta @ Home Aide à la prédiction de structures et des complexes protéiques.
- Le repliement des protéines & l’I.A AlphaFold de DeepMind – Science étonnante de David Louapre.