

| Spécialité | Neurologie |
|---|
| DiseasesDB | 8358 |
|---|---|
| MeSH | D016472 |
Les maladies des motoneurones (MND) sont un groupe de troubles neurodégénératifs qui affectent sélectivement les motoneurones, les cellules qui contrôlent les muscles volontaires du corps.
Selon la CIM-11, les troubles suivants sont comptés parmi les maladies des motoneurones: la sclérose latérale amyotrophique (SLA), la paralysie bulbaire progressive (PBP), la paralysie pseudobulbaire, l'atrophie musculaire progressive (PMA), la sclérose latérale primaire (PLS) et l' amyotrophie monomélique (MMA), ainsi que certaines variantes plus rares ressemblant à la SLA.
Les maladies des motoneurones affectent à la fois les enfants et les adultes. Bien que chaque maladie du motoneurone affecte les patients différemment, elles provoquent toutes des symptômes liés aux mouvements, principalement une faiblesse musculaire. La plupart de ces maladies semblent se produire au hasard sans causes connues, mais certaines formes sont héréditaires. Des études sur ces formes héréditaires ont conduit à la découverte de divers gènes (par ex. SOD1) qui sont jugées importantes pour comprendre comment la maladie survient.
Les symptômes des maladies des motoneurones peuvent être observés pour la première fois à la naissance ou peuvent apparaître lentement plus tard dans la vie. La plupart de ces maladies s'aggravent avec le temps; tandis que certains raccourcissent l’espérance de vie (par ex. ALS), d'autres non.
Actuellement, il n'existe aucun traitement approuvé pour la majorité des troubles des motoneurones, et les soins sont principalement symptomatiques.
Signes et symptômes
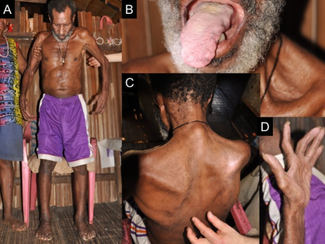
Les signes et les symptômes dépendent de la maladie spécifique, mais les maladies des motoneurones se manifestent généralement sous la forme d'un groupe de symptômes liés aux mouvements. Ils progressent lentement et s'aggravent en plus de trois mois. Divers schémas de faiblesse musculaire sont observés et des crampes et des spasmes musculaires peuvent survenir. On peut avoir des difficultés à respirer lors de la montée des escaliers (effort), des difficultés à respirer en position couchée (orthopnée), ou même une insuffisance respiratoire si les muscles respiratoires sont impliqués. Des symptômes bulbaires, notamment des difficultés à parler (dysarthrie), des difficultés à avaler (dysphagie) et une production excessive de salive (sialorrhée), peuvent également survenir. La sensation, ou la capacité de ressentir, n'est généralement pas affectée. Des troubles émotionnels (par exemple, un effet pseudobulbaire) et des changements cognitifs et comportementaux (par exemple, des problèmes de fluidité des mots, de prise de décision et de mémoire) sont également observés. Il peut y avoir des anomalies des motoneurones (par exemple, fonte musculaire, contractions musculaires), des anomalies des motoneurones (par exemple, réflexes rapides, réflexe de Babinski, réflexe de Hoffman, augmentation du tonus musculaire), ou les deux.
Les maladies des motoneurones sont observées tant chez les enfants que chez les adultes. Ceux qui affectent les enfants ont tendance à être héréditaires ou familiaux, et leurs symptômes sont présents à la naissance ou apparaissent avant d'apprendre à marcher. Ceux qui affectent les adultes ont tendance à apparaître après 40 ans. L'évolution clinique dépend de la maladie spécifique, mais la plupart progressent ou s'aggravent au cours des mois. Certains sont mortels (par exemple ALS), tandis que d'autres ne le sont pas (par ex. PLS).
Modèles de faiblesse
Divers modèles de faiblesse musculaire se produisent dans différentes maladies des motoneurones. La faiblesse peut être symétrique ou asymétrique, et elle peut se produire dans les parties du corps distales, proximales ou les deux. . . Selon Statland et al., Il existe trois principaux schémas de faiblesse observés dans les maladies des motoneurones, qui sont :
- Faiblesse distale asymétrique sans perte sensorielle (par ex. ALS, PLS, PMA, MMA)
- Faiblesse symétrique sans perte sensorielle (par ex. PMA, PLS)
- Faiblesse proximale médiane focale symétrique (cou, tronc, atteinte bulbaire; par ex. ALS, PBP, PLS)
Découvertes des neurones moteurs inférieurs et supérieurs
Les maladies des motoneurones sont sur un spectre en termes d'implication des motoneurones supérieurs et inférieurs. Certains ont juste des résultats de motoneurones inférieurs ou supérieurs, tandis que d'autres ont un mélange des deux. Les découvertes des motoneurones inférieurs (LMN) incluent l'atrophie musculaire et les fasciculations, et les découvertes des motoneurones supérieurs (UMN) incluent l'hyperréflexie, la spasticité, les spasmes musculaires et les réflexes anormaux.
Les maladies du motoneurone pur ou celles qui ne présentent que des UMN incluent le PLS.
Les maladies des neurones moteurs purs ou celles qui ne présentent que des LMN incluent le PMA.
Les maladies des motoneurones avec des résultats à la fois UMN et LMN incluent la SLA familiale et sporadique.
Les causes
La plupart des cas sont sporadiques et leurs causes ne sont généralement pas connues. On pense que des facteurs environnementaux, toxiques, viraux ou génétiques peuvent être impliqués.
Dommages à l'ADN
TARDBP (TAR DNA-binding protein 43), également appelé TDP-43, est un composant essentiel de la voie enzymatique non homologue de la jonction d'extrémité (NHEJ) qui répare les cassures double brin de l'ADN dans les motoneurones pluripotents dérivés des cellules souches. Le TDP-43 est rapidement recruté pour des ruptures double brin où il agit comme un échafaudage pour le recrutement du complexe protéine XRCC4 - ADN ligase qui agit ensuite pour réparer les ruptures double brin. Environ 95% des patients SLA ont des anomalies dans la localisation cytoplasmique du TDP43 dans les motoneurones spinaux du noyau. Dans les motoneurones dérivés de cellules souches neurales humaines appauvries en TDP-43, ainsi que dans les échantillons de moelle épinière des patients atteints de SLA sporadique, il y a une accumulation significative de cassure double brin et des niveaux réduits de NHEJ.
Facteurs de risque associés
Chez les adultes, les hommes sont plus souvent touchés que les femmes.
Diagnostic
Le diagnostic différentiel peut être difficile en raison du nombre de symptômes qui se chevauchent, partagés entre plusieurs maladies des motoneurones. Souvent, le diagnostic est basé sur des résultats cliniques (c.-à-d. LMN vs. Signes et symptômes UMN, schémas de faiblesse), antécédents familiaux de MND, et une variété de tests, dont beaucoup sont utilisés pour exclure les mimiques de la maladie, qui peuvent se manifester avec des symptômes identiques.
Veuillez vous référer aux articles individuels pour les méthodes de diagnostic utilisées dans chaque maladie individuelle des motoneurones.
Classification
La maladie des motoneurones décrit une collection de troubles cliniques, caractérisés par une faiblesse musculaire progressive et la dégénérescence du motoneurone lors des tests électrophysiologiques. Comme discuté ci-dessus, le terme «maladie des motoneurones» a différentes significations dans différents pays. De même, la littérature classe de manière incohérente les troubles dégénératifs des motoneurones qui peuvent être inclus sous le terme générique de "maladie des motoneurones". Les quatre principaux types de MND sont marqués (*) dans le tableau ci-dessous.
Tous les types de MND peuvent être différenciés par deux caractéristiques déterminantes :
- La maladie est-elle sporadique ou héréditaire?
- Y a-t-il une implication des motoneurones supérieurs (UMN), des motoneurones inférieurs (LMN), ou des deux?
Des MND sporadiques ou acquis surviennent chez des patients sans antécédents familiaux de maladie dégénérative des motoneurones. Les MND héréditaires ou génétiques adhèrent à l'un des modèles d'hérédité suivants: autosomique dominant, autosomique récessif ou lié à l'X. Certains troubles, comme la SLA, peuvent survenir sporadiquement (85%) ou peuvent avoir une cause génétique (15%) avec les mêmes symptômes cliniques et la même progression de la maladie.
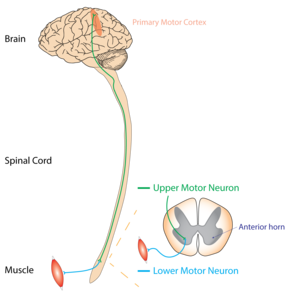
Les UMN sont des motoneurones qui se projettent du cortex jusqu'au tronc cérébral ou à la moelle épinière. Les LMN trouvent leur origine dans les cornes antérieures de la moelle épinière et se synaptisent sur les muscles périphériques. Les deux neurones moteurs sont nécessaires à la forte contraction d'un muscle, mais les dommages à un UMN peuvent être distingués des dommages à un LMN par un examen physique.
| Type | Dégénérescence UMN | Dégénérescence LMN |
|---|---|---|
| MND sporadiques | ||
| Sclérose latérale amyotrophique sporadique (SLA) * | Oui | Oui |
| Sclérose latérale primaire (PLS) * | Oui | Non |
| Atrophie musculaire progressive (PMA) * | Non | Oui |
| Paralysie bulbaire progressive (PBP) * | Oui | Oui, région bulbaire |
| Paralysie pseudobulbaire | Oui, région bulbaire | Non |
| Amyotrophie monomélique (MMA) | Non | Oui |
| MND hérités | ||
| Sclérose latérale amyotrophique familiale (SLA) * | Oui | Oui |
Les tests
- Tests de liquide céphalo-rachidien (LCR): l'analyse du liquide provenant du cerveau et de la moelle épinière pourrait révéler des signes d'infection ou d'inflammation.
- Imagerie par résonance magnétique (IRM): une IRM du cerveau et de la moelle épinière est recommandée chez les patients présentant des signes et symptômes UMN pour explorer d'autres causes, telles qu'une tumeur, une inflammation ou un manque d'approvisionnement en sang (accident vasculaire cérébral).
-
Électromyogramme (EMG) et étude de conduction nerveuse (NCS): l'EMG, qui évalue la fonction musculaire, et le NCS, qui évalue la fonction nerveuse, sont réalisés ensemble chez les patients présentant des signes LMN.
- Pour les patients atteints de MND affectant les LMN, l'EMG montrera des preuves de: (1) dénervation aiguë, qui se poursuit à mesure que les motoneurones dégénèrent, et (2) dénervation et réinnervation chronique du muscle, alors que les motoneurones restants tentent de se remplir. pour les motoneurones perdus.
- En revanche, le SNC chez ces patients est généralement normal. Il peut montrer un faible potentiel d'action musculaire composé (CMAP), qui résulte de la perte de motoneurones, mais les neurones sensoriels ne devraient pas être affectés.
- Biopsie tissulaire : La prise d'un petit échantillon d'un muscle ou d'un nerf peut être nécessaire si l'EMG / NCS n'est pas suffisamment spécifique pour exclure d'autres causes de faiblesse musculaire progressive, mais il est rarement utilisé.
Traitement
Il n'existe aucun traitement curatif connu pour la majorité des troubles des motoneurones. Veuillez vous référer aux articles sur les troubles individuels pour plus de détails.
Pronostic
Le tableau ci-dessous répertorie l'espérance de vie des patients qui reçoivent un diagnostic de MND. Veuillez vous référer aux articles individuels pour plus de détails.
| Type | Temps de survie médian dès le début des symptômes |
|---|---|
| Sclérose latérale amyotrophique (SLA) | 2 à 5 ans |
| Sclérose latérale primaire (PLS) | 8 à 10 ans |
| Atrophie musculaire progressive (PMA) | 2 à 4 ans |
| Paralysie bulbaire progressive (PBP) | 6 mois - 3 ans |
| Paralysie pseudobulbaire | Aucun changement dans la survie |
Terminologie
Aux États-Unis, le terme maladie des motoneurones est souvent utilisé pour désigner la sclérose latérale amyotrophique (maladie de Lou Gehrig), le trouble le plus courant du groupe. Au Royaume-Uni, le terme est une maladie des motoneurones orthographiée et est fréquemment utilisé pour l'ensemble du groupe mais peut également désigner spécifiquement la SLA.
Alors que MND se réfère à un sous-ensemble spécifique de maladies similaires, il existe de nombreuses autres maladies des motoneurones qui sont appelées collectivement "troubles du motoneurone", par exemple les maladies appartenant au groupe des atrophies musculaires spinales. Cependant, ils ne sont pas classés comme «maladies des motoneurones» par la 11e édition de la Classification statistique internationale des maladies et des problèmes de santé connexes (CIM-11) qui est la définition suivie dans cet article.
Voir également
- Atrophies musculaires spinales
- Neuropathies motrices et sensorielles héréditaires