
| Symptômes | Enchondrome (en) et enchondromatose (en) |
|---|
| Spécialité | Génétique médicale |
|---|
| Prévalence | 1/100 000 |
|---|
| CIM-10 | Q78.4 |
|---|---|
| CIM-9 | 756.4 |
| OMIM | 166000 |
| DiseasesDB | 9212 |
| MedlinePlus | https://medlineplus.gov/genetics/condition/ollier-disease/ |
| MeSH | D004687 |
La maladie d'Ollier ou enchondromatose est une maladie constitutionnelle de l'os non héréditaire et sporadique, dans laquelle se développe des tumeurs cartilagineuses bénignes, les enchondromes. Ces derniers peuvent être de nombre, de taille, et de localisations différents expliquant l'extrême variabilité clinique entre les porteurs de cette maladie. Les principaux troubles observables sont une asymétrie et un raccourcissement des membres touchés, avec des déformations. Les symptômes apparaissent généralement pendant la petite enfance, lors de la croissance. Les atteintes sont généralement prédominantes d'un côté.
De nombreux patients atteints par la maladie d'Ollier sont susceptibles de développer des tumeurs malignes, en particulier des sarcomes osseux de type chondrosarcome, mais également des tumeurs dans d'autres tissus.
Un lien a été démontré entre la maladie d'Ollier et des mutations sur les gênes IDH1, IDH2 et PTH1R.
Actuellement, il n'existe aucun traitement de fond pour la maladie d'Ollier, mais les complications telles que les fractures, différences de longueur, ou tumeurs malignes agressives peuvent être traitées par chirurgie et/ou thérapie adjuvante.
L'association de la maladie d'Ollier à des hémangiomes prend le nom de syndrome de Maffucci.
Historique et épidémiologie
Connue depuis longtemps, ses caractéristiques ont été définies en 1899 par le chirurgien Louis Léopold Ollier qui lui donna son nom.
Sa prévalence est estimée à 1 cas sur 100 000 individus.
Cette maladie fait partie des maladies rares (117 personnes recensées en France en 2017 par l'Association Ollier-Maffucci Europe).
Synonymes : dyschondroplasie, hémichondrodystrophie d'Ollier, hémichondrodysplasie, chondromatose enchondrale multiple, syndrome d'Ollier-Mollin.
Diagnostic
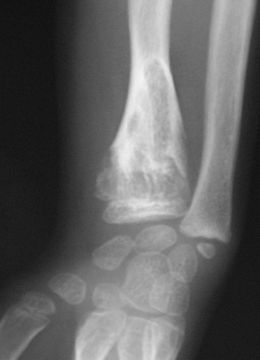
Les premières manifestations de la maladie apparaissent en général durant la petite enfance du fait d'un développement perturbé de l'os par la croissance d'enchondromes multiples entraînant déformations, déviations axiales et différences de longueur entre les membres. Les tuméfactions osseuses se présentent radiologiquement comme des lacunes claires arrondies, finissant par se calcifier. La pose du diagnostic se fait par un examen radiologique.
Complications associées
La principale complication de la maladie d'Ollier est la transformation sarcomateuse d'un chondrome en chondrosarcome. Cette transformation est estimée, selon les études, en général entre 25 et 30% mais pouvant aller jusqu'à 50%. Bien que le chondrosarcome soit la forme la plus courante de tumeur osseuse maligne secondaire dans les cas de maladies d'Ollier, d'autres formes comme le chordome et l'ostéosarcome peuvent se produire.
La maladie d'Ollier comporte également un risque élevé de favoriser le développement de tumeurs malignes agressives dans d'autres tissus tel que le cerveau, les ovaires, ou le foie. Il a été également rapporté des cas de maladie d'Ollier associés à des leucémies ou myélodysplasies. Une majorité de ces tumeurs comportent des mutations sur les gênes IDH1 et IDH2, pouvant expliquer le lien avec la maladie.
Non traitées, ces transformations malignes peuvent entraîner des conséquences fatales.
Traitement et suivi
Il n'existe pas de traitement de fond, la prise en charge est uniquement symptomatique. Selon le degré d'atteinte, les traitements seront essentiellement correctifs, tels que des allongement de membres (Illizarov/Fixateur externe), couplés à une prise en charge de la douleur. Les atteintes des chondromes évoluant avec la croissance, les effets de la maladie se stabilisent à la fin de la puberté, mais un suivi adulte régulier reste indispensable pour dépister précocement toutes formes de complications associées.
A l'âge adulte, le suivi clinique et radiologique sont les principaux outils pour dépister la survenue de transformation sarcomateuse des chondromes.
S'agissant du risque sur les autres tissus, il est recommandé d'effectuer un bilan complet (IRM cérébral, échographie hépatique, Imagerie gastro-intestinale) régulièrement.
Causes
Pendant de nombreuses années, la plupart des recherches n'ont pu identifier la cause de la maladie.
Les études récentes ont démontré que la plupart des cas (81%) de maladie d'Ollier sont causés par des mutations de l'isocitrate déshydrogénase 1 et 2 (IDH1 et IDH2). IDH1 et IDH2 sont normalement des enzymes qui catalysent la conversion de l'isocitrate en α-cétoglutarate. Les formes mutées d'IDH1 et d'IDH2 entraînent par la suite une conversion de l'α-cétoglutarate en 2-hydroxyglurate. La surproduction de 2-hydroxyglurate a pour effet d'inhiber la différenciation ostéogénique, tout en stimulant la différenciation chondrogénique, expliquant le développement des enchondromes.
Environ 8 à 10% des cas de maladie d'Ollier sont liés à une sur PTH1R.
Vu la distribution des atteintes sur un individu, il est hautement probable que la maladie résulte de mutations somatiques post-zygotique résultant un trouble génétique mosaïque.
Transmission
La maladie d'Ollier n'est pas transmissible et n'est pas une maladie héréditaire.
Les cas sont sporadiques et confirment la non-transmission mendélienne de cette maladie.
Voir aussi
Références
Liens externes
- Association Ollier Maffucci Europe
- « Ollier disease », Orphanet Journal of Rare Diseases, vol. 1, no 37, (DOI 10.1186/1750-1172-1-37, lire en ligne)