
| Spécialité | Oncologie |
|---|
| CIM-10 | C96.6 |
|---|---|
| ICD-O | 9752/3, 9751/1 et 9751/3 |
| OMIM | et 246400 604856 et 246400 |
| DiseasesDB | 5906 |
| eMedicine | 1100579 |
| MeSH | D006646 |

L'histiocytose langerhansienne (anciennement histiocytose X) est une maladie orpheline caractérisée par une accumulation de macrophages (ou histiocytes) dans les tissus. Ces histiocytes présentent la caractéristique de cellules de Langerhans. L'histiocytose langerhansienne est causée par un processus néoplasique clonal.
Historique
Données médicales
Les premières descriptions de la maladie sont faites de 1893 à 1919, et réunies sous le nom de maladie de Hand-Shuller-Christian, une maladie rare du jeune enfant associant une exophtalmie, un diabète insipide et des lésions crâniennes (lacunes osseuses dues à des granulomes à éosinophiles). Ce n'était qu'une des formes (atteinte hypophysaire) de la maladie.
Par la suite, une autre maladie rare du nourrisson est décrite sous le nom de maladie de Letterer-Siwe, elle se manifeste comme une maladie systémique, le plus souvent aiguë et d'évolution fatale : atteintes cutanées et hépatiques, infiltration de la moelle osseuse par des cellules réticulo-endothéliales. D'autres variantes moins graves seront décrites comme la forme cutanéo-pulmonaire (réticulose de Julien Marie) en 1941, ou la forme cutanée exclusive (histiocytose de Hashimoto-Prizker).
La terminologie s'est simplifiée en 1953 grâce à deux pathologistes, Farber et Lichtenstein qui unifient ces différentes manifestations en une seule affection dite histiocytose X (le X indiquant l'inconnue de la cause ou de l'origine de l'infiltration par des histiocytes atypiques).
Dans les années 1960, les travaux en microscope électronique de Françoise Basset permettent à Christian Nezelof de rapprocher ces histiocytes de formes pathologiques de cellules de Langerhans . Ces travaux sont confirmés, et en 1987, l'histiocytisis X est renommée internationalement Langerhans cell histiocytosis ou Histiocytose langerhansienne.
Paléopathologie
Le Cro-Magnon 1 , l'un des premiers sapiens (homme adulte) découvert en France en 1868, présentait une érosion frontale et des lésions de la mandibule gauche, qui ont fait l'objet d'un grand nombre de diagnostics rétrospectifs. En 1981, ces lésions (retrouvées aussi sur d'autres parties du squelette) ont été identifiées comme compatibles avec une histiocytose X.
D'autres cas probables sont ceux d'un enfant âgé de 2 à 3 ans, sur un site de l'Illinois daté de 1000-1600 ap. J.C, et d'une jeune femme du néolithique en Espagne.
Causes
L'histiocytose langerhansienne est un processus néoplasique clonal, lié notamment à des mutations de gènes de la voie des MAP-kinase, à l'instar du gène BRAF. La découverte de ces mutations date de 2010 et a permis de définitivement prouver son caractère clonale. Avant cela, l'histiocytose langerhansienne, bien que se manifestant par une prolifération/infiltration de cellules atypiques dans les tissus, n'apparaissait pas liée à une cause définie et les conceptions réactionnelle et clonale s'opposaient.
Dans la conception réactionnelle, l'accent est mis sur des processus immunitaires déclenchés contre un antigène inconnu (agent extérieur). Dans la conception tumorale, c'est le caractère clonal de la prolifération cellulaire, perturbée par des mutations génétiques, qui est mis en avant. Ces deux approches ne sont pas incompatibles, et des auteurs estiment que l'histiocytose réalise une combinaison des deux. Il est important de considérer qu'en dépit du caractére clonal de l'histiocytose langerhansienne, cette maladie ne possède pas les caractéristiques communes des cancers, comme la prolifération cellulaire, l'extension locale ou régionale, l'évolution vers des métastastases et là aussi de façon différentes des cancers, possède une capacité de guérison spontanée.
Dans la très grande majorité des cas, aucune cause n'est trouvée. On considère que cette maladie est liée à la fois à une susceptibilité individuelle et à certaines stimulations virales ou « chimiques ». Ainsi, le lien entre la forme pulmonaire de la maladie et le tabagisme est solidement établi (plus de 90% des patients adultes sont fumeurs), mais parmi les fumeurs, l'incidence des histiocytoses est très faible. Il y aurait donc un terrain particulier pour déclencher une atteinte pulmonaire en cas de contact avec le tabac (quelques rares cas familiaux ont été rapportés).
Le rôle de virus a été suggéré, comme celui du herpèsvirus humain type 6, mais infirmé par d'autres études.
Épidémiologie
L'histiocytose langerhansienne est une maladie rare, mais elle peut atteindre toute tranche d'âge, du nourrisson à la personne âgée. Il existe une incidence nettement plus élevé de la naissance à 3 ans. En Europe, son incidence chez l'adulte est estimée à 1 ou 2 nouveaux cas par million, il s'agit d'une sous-estimation car les adultes sont sous-diagnostiqués.
En France, chez l'enfant de moins de 15 ans, l'étude réalisée sur la période 2000–2004 donne 4,6 cas par million, soit 55 nouveaux cas par an. L’incidence était plus élevée chez les nourrissons (15,3 cas par million), et diminuait avec l’âge pour atteindre 2 par million après 10 ans.
Aux États-Unis, des études américaines intégrant des données ethniques indiquent que la maladie est plus fréquente chez les enfants hispaniques, et moins fréquente chez les enfants noirs.
Clinique
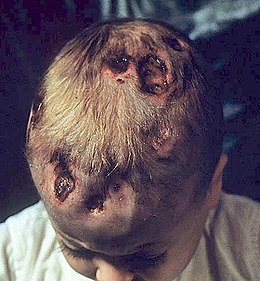
La maladie se manifeste par des poussées de durée et de gravité variables, allant d'une atteinte unique à des formes progressives touchant plusieurs organes vitaux. Les atteintes de la maladie, très variables, peuvent aller de lésions cutanées locales à la dégradation de divers tissus. L'os est souvent atteint. Les formes graves peuvent toucher plusieurs organes vitaux (moelle osseuse, foie, poumon). La maladie peut aussi atteindre l'hypophyse (et donc être responsable de déficit hormonal en premier le diabète insipide central par déficit de l'hormone anti diurétique), plus exceptionnellement, du système nerveux.
Chez l'enfant
Classiquement, on distingue la forme du nourrisson et celle de l'enfant plus grand ou de l'adolescent. Les formes du nourrisson sont le plus souvent multifocales. En pratique, il existe de nombreuses formes intermédiaires, et qui peuvent aussi se voir chez les personnes âgées.
Atteinte systémique de la maladie avec atteinte des organes à risque (ici= atteinte du foie, de la rate, hématologique). Il existe de rares formes congénitales, mais cette affection touche les plus souvent le nourrisson entre le 3e et le 6e mois, rarement après le 18e. L'atteinte cutanée est ici très fréquente faite d'une éruption de papules croûteuse et d'un purpura, touchant la tête et le tronc, mais respectant les membres. D'autres manifestations cutanées sont plus rares. Quand cette forme cutanée reste isolée, l'évolution est bénigne. Mais l'association avec une atteinte du foie, de la rate ou une cytopénie (anémie et baisse des plaquettes) mettent ici en jeu le pronostic vital.
Chez l'enfant plus âgé que 2 ans, la maladie est en règle générale moins grave. L'atteinte la plus fréquente est l'atteinte osseuse, qui concerne d'abord les os du crâne, les vertèbres. Les localisations osseuses, pulmonaires et ganglionnaires sont possibles. La triade historique (exophtalmie, diabète insipide, lacunes crâniennes) se rencontre rarement. L'évolution est variable et le pronostic dépend de l'importance des conséquences fonctionnelles de chaque atteinte. Une atteinte de la voute du crane peut se limiter à une tuméfaction transitoire, mais une atteinte de l'os temporal peut menacer l'audition.
Chez l'adulte
Considérée comme exceptionnelle à la fin du XXe siècle (environ 80 cas publiés dans la littérature mondiale), la forme de l'adulte apparait comme sous-diagnostiquée au début du XXIe siècle . Par rapport à l'enfant, elle ne touche le plus souvent qu'un seul tissu, avec moins de localisations.
L'atteinte pulmonaire isolée survient essentiellement chez l'adulte jeune. La maladie est asymptomatique ou non spécifique (toux, dyspnée, altérations de l'état général). Dans 10 à 20% des cas, la maladie est découverte à l'occasion d'un pneumothorax spontané. La radiographie montre des micronodules répartis symétriquement dans les deux poumons (partie haute et moyenne), où l'on peut individualiser des kystes. La structure et l'évolution de ces kystes sont précisées par la tomodensitométrie pulmonaire. Le diagnostic de certitude repose sur la biopsie chirurgicale guidée par l'imagerie, dont la nécessité est discutée au cas par cas.
Bilan
Ces différentes distinctions cliniques tendent à être supplantées par de nouvelles classifications, dont celles de l'Histiocyte Society (société savante internationale sur la maladie). L'une distingue 4 groupes de maladies : 1) maladie mono-systémique (uni- ou multifocale), 2) maladie multi-systémique, 3) maladie multi-systémique avec atteinte d’organe à risque. Enfin l'atteinte pulmonaire, quoique non considérée comme 'à risque' est malgré tout potentiellement sévère et se distingue. Une autre, en cours d'élaboration pourrait se baser sur la génomique, à partir des mutations détectées dans la production d'histiocytes par la moelle osseuse (cadre général de l'hématopoièse).
Dans les années 2010, de réels progrès biologiques et génomiques ont permis d'avancer dans la compréhension de cette maladie (trouble de la différenciation myéloïde des histiocytes). Mais le défi demeure de transformer ces avancées en améliorations concrètes pour les malades, enfants comme adultes.
Est-ce un cancer ?
L'histiocytose langerhansienne n'est pas une maladie génétique héréditaire et n'est pas contagieuse. La frontière de cette maladie avec le cancer est parfois difficile à comprendre mais certaine.
L'histiocytose langerhansienne est parfois très sévère et peut mettre en jeu la vie de la personne. De plus quand un traitement est mis en place, il s'agit de chimiothérapie et donc le lieu de soins est souvent un service de cancérologie. Sur le plan biologique, il est maintenant certain que dans le tissu pathologique d'environ la moitié des cas, il existe une anomalie clonale avec mutation d'un gène de la voie des MAP-kinases, à l’instar du gène BRAF. Enfin, une faible proportion des patients présentent dans leur vie à la fois une histiocytose langerhansienne et un cancer. Tous ces faits plaideraient pour considérer que l'histiocytose langerhansienne est un cancer.
Les arguments contre sont le pronostic le plus souvent favorable, le principe d'utilisation des chimiothérapies et la biologie.
Dans près de 50 % des cas, la maladie guérit spontanément en l'absence de traitement. Le principe du traitement n'est pas d'éliminer un clone malin, comme dans un cancer, mais plutôt de limiter la réaction inflammatoire et la destruction des tissus. L'évolution de la maladie est enfin marquée par des poussées parfois sur plusieurs organes différents et non pas par un processus de métastases. La justification de la prise en charge des patients en cancérologie ou hématologie, n'est pas qu'il s'agisse d'un cancer. Il s'agit d'une question d'organisation des soins. En effet, si une chimiothérapie est prescrite, elle doit être administrée par une équipe qualifiée en hémato-oncologie. Ceci est toujours le cas en pédiatrie, et pour l'adulte, les services de médecine interne peuvent aussi administrer ces traitements. Le dernier point est biologique, car l'oncogène B-raf n'a pas obligatoirement un effet oncogénique et peut être retrouvé en dehors de toute prolifération maligne (comme dans les nævus ou « grains de beauté »).
Traitements
Les atteintes localisées ne demandent souvent qu'une prise en charge limitée. Les formes touchant des organes vitaux nécessitent des traitements lourds : chimiothérapie, corticothérapie, et thérapie ciblée dans certains cas.
La réponse aux traitements n'est pas toujours prévisible : la maladie peut continuer d'évoluer, régresser ou disparaître et, dans certains cas, récidiver.
L'arrêt du tabac est impératif.
Lieux de soins
En France, les lieux de soins peuvent être accessibles à travers le site www.eurohistio.net qui offre une carte des centres experts. Schématiquement, la maladie est diagnostiquée par de nombreux spécialistes aussi différents que les orthopédistes, les stomatologues, les dermatologues, les endocrinologues, les hématologues, les oncologues, les pneumologues, les internistes, les rhumatologues, les neurochirugiens, les neurologues, les hépatologues, les radiologues, tandis que la preuve est donnée par un médecin anatomopathologiste… Le soin peut être assuré par le spécialiste qui fait le diagnostic de la maladie, mais il est fortement recommandé qu'un contact soit pris avec le centre de référence pour assurer la cohérence de la prise en charge globale du patient et pour avoir la meilleure décision thérapeutique. Le centre de référence est composé par plusieurs services hospitaliers sur Paris et en province. L'équipe du centre de référence peut voir les patients de toute la France, mais en règle le projet de soin est discuté avec le médecin traitant. Plusieurs services, quoique ne composant pas le centre de référence, sont très actifs et coopérant. Ceci est en particulier le cas des centres pédiatriques d'hématologie et oncologie. Une fois par mois, une réunion a lieu pour aider à l'expertise des cas, si des questions se posent. L'expertise est ainsi assurée la plupart du temps, sans que le patient ait à se déplacer.
Registre des patients
Un registre des patients a été mis en place en France et est localisé à l'hôpital Trousseau, Paris. Ce registre, labellisé par le comité national des registres, et déclaré à la CNIL, assure la surveillance épidémiologique et offre des informations sur le suivi des patients.
Liens externes
- le site de l'association des patients français et du réseau français des médecins avec contact auprès du centre de référence :
- le site EU des centres de référence histiocytose - en 8 langues dont le français
- le site de l'Histiocyte Society, société savante internationale
- : un petit livret pour expliquer la maladie à un enfant