
| CIM-10 | E71.3 |
|---|---|
| CIM-9 | 277.85 |
| OMIM | 201470 |
| DiseasesDB | 31599 |
| MeSH | C537596 |
Le déficit en acyl-coenzyme A déshydrogénase defs acides gras à chaîne courte (SCADD), également appelé déficit en ACADS ou déficit en SCAD, est une maladie autosomique récessive d'oxydation des acides gras trouble qui affecte les enzymes nécessaires à briser un certain groupe de graisses appelées acides gras à courte chaîne.
Signes/symptômes
Les nourrissons atteints de SCADD auront des vomissements, un taux de sucre dans le sang faible, un manque d'énergie (léthargie), une mauvaise alimentation, et l'absence de gain de poids et de croissance. D'autres caractéristiques de ce trouble peuvent inclure un manque de tonus musculaire (hypotonie), des convulsions, des retards de développement et une microcéphalie. Les symptômes de SCADD peuvent apparaître au cours de maladies telles que les infections virales. Dans certains cas, les signes et les symptômes peuvent ne pas apparaître jusqu'à l'âge adulte, lorsque certaines personnes peuvent développer des faiblesses musculaires, tandis que d'autres personnes auront des symptômes légers et ne seront jamais diagnostiquées.
La génétique
La SCADD est causée génétiquement par des mutations dans les gènes ACADS, localisé sur le chromosome 12q22-qter. Les mutations dans les gènes ACADS conduisent à l'insuffisance des niveaux d'acyl-CoA déshydrogénase à chaîne courte, ce qui est important pour briser les chaînes courtes d'acides gras. De faibles niveaux de cette enzyme stoppe le cycle de décomposition et de transformation des acides gras à chaîne courte dans les mitochondries. Par conséquent, ces acides gras à chaîne courte seront pas convertis en énergie.
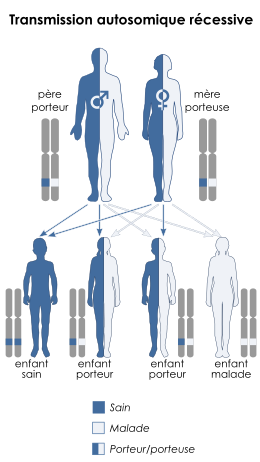
Le trouble est héritée par transmission autosomique récessive. Cela signifie que le gène défectueux responsable de la maladie est situé sur un autosome (le chromosome 12 est un autosome), et que deux copies du gène défectueux sont nécessaires afin d'être porteur atteint de cette maladie. Les parents d'un individu avec une maladie autosomique récessive sont tous les deux porteur d'une copie défectueuse du gène.
Diagnostic
Le diagnostic de courte chaîne acyl-coenzyme A déshydrogénase est basé sur les éléments suivants:
Traitement
En termes de traitement pour la SCADD, certaines personnes peuvent ne pas avoir besoin de traitement, tandis que d'autres pourraient suivre l'administration de:
Épidémiologie
Ce trouble, du point de vue épidémiologique, affecte environ 1 nouveau-né sur 50 000 selon Jethva, et al. Cependant, dans l'état américain de Californie, il semble y avoir un rapport de 1 pour 35 000.
Références
Lectures complémentaires
- (en) Loren Pena, Brad Angle, Barbara Burton et Joel Charrow, « Follow-up of patients with short-chain acyl-CoA dehydrogenase and isobutyryl-CoA dehydrogenase deficiencies identified through newborn screening: one center's experience », Genetics in Medicine, vol. 14, no 3, , p. 342–347 (ISSN 1098-3600, DOI 10.1038/gim.2011.9, lire en ligne, consulté le )
- (en) John Fernandes, Jean-Marie Saudubray, Georges van den Berghe et John H. Walter, Inborn Metabolic Diseases : Diagnosis and Treatment, Berlin, Springer Science & Business Media, (ISBN 978-3-540-28785-8, lire en ligne)